






在生命科学研究里,“蛋白质组学”就像解读生命密码的钥匙——它研究蛋白质及其翻译过程、亚型组成、翻译后修饰(PTM)和降解机制,揭开疾病发生、细胞活动的底层逻辑。在自下而上的蛋白质组学研究中,解决肽段复杂性是基于质谱(MS)的蛋白质组学数据采集和生物信息学分析的核心问题。相比数据依赖采集(DDA),数据非依赖采集(DIA)采用相对较宽的隔离窗口,无偏倚地采集窗口内的所有肽段,动态范围大大提高,实现了更高深度的蛋白质组鉴定,成为单细胞蛋白质组、大规模样本分析的“主力军”。
DIA虽好,却一直有三座“大山”让人头疼
♦ 数据复杂难处理:新一代TOF检测器能捕捉到 “单分子级” 信号,但原始数据里全是无清晰峰形的 “杂乱信号”,传统方法会弄丢关键信息;
♦ 建库太麻烦:以前做DIA得先做DDA预实验,建 “样本专属经验谱库”,耗时耗力还没法通用;
♦ 工具不灵活:主流分析工具多是闭源的,遇到新型仪器(如timsTOF)或未知蛋白质修饰(PTM),直接 “水土不服”。
2025年10月21日,针对DIA分析的“老大难”,Matthias Mann团队在《Nature Biotechnology》发表了题为“AlphaDIA enables DIA transfer learning for feature-free proteomics”的文章,推出开源的AlphaDIA的搜库软件,用“无特征处理 + 深度学习迁移学习”的方式搬平DIA数据解析的三座大山。

一、AlphaDIA的工作流程
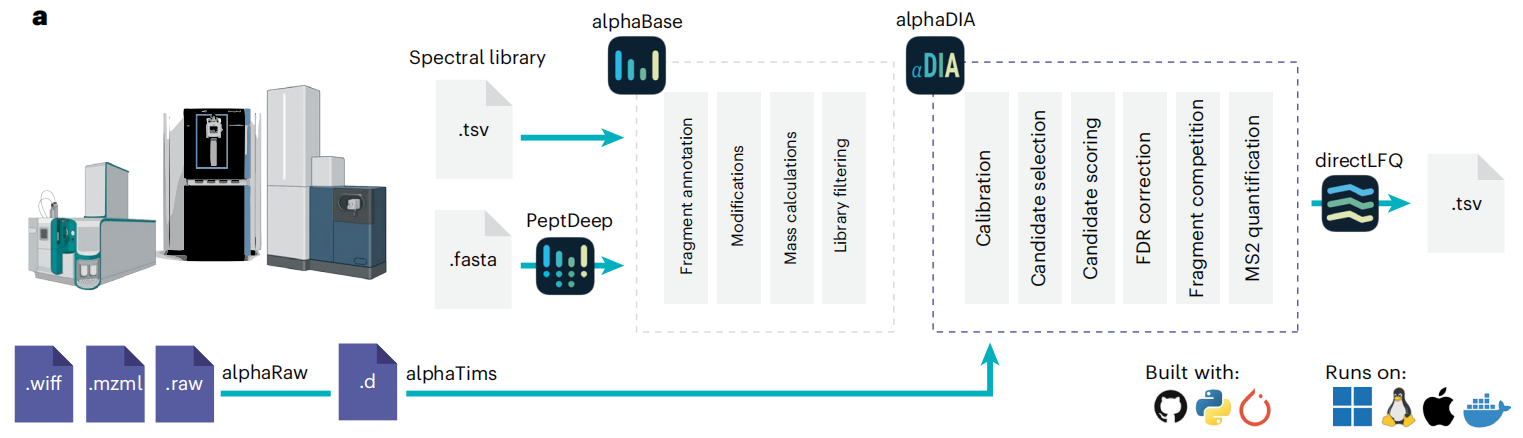
Figure 1
AlphaDIA是一款用于DIA搜索的模块化开源框架。该框架基于科学Python工具栈和 alphaPept生态系统构建,支持灵活的搜索策略,可通过Python API、Jupyter notebooks、命令行界面或易于安装的图形用户界面访问。AlphaDIA涵盖了从原始文件到蛋白定量报告的完整工作流程,能够处理所有主流厂商的文件及专有格式。其设计初衷是实现大型队列的“一站式处理”,可在Windows、Linux和Mac系统上原生运行,也能通过Slurm 或Docker以分布式方式在云端部署。
二、AlphaDIA的三大核心突破,每一个都很能打
突破1:“无特征处理”,让TOF数据不再“浪费”
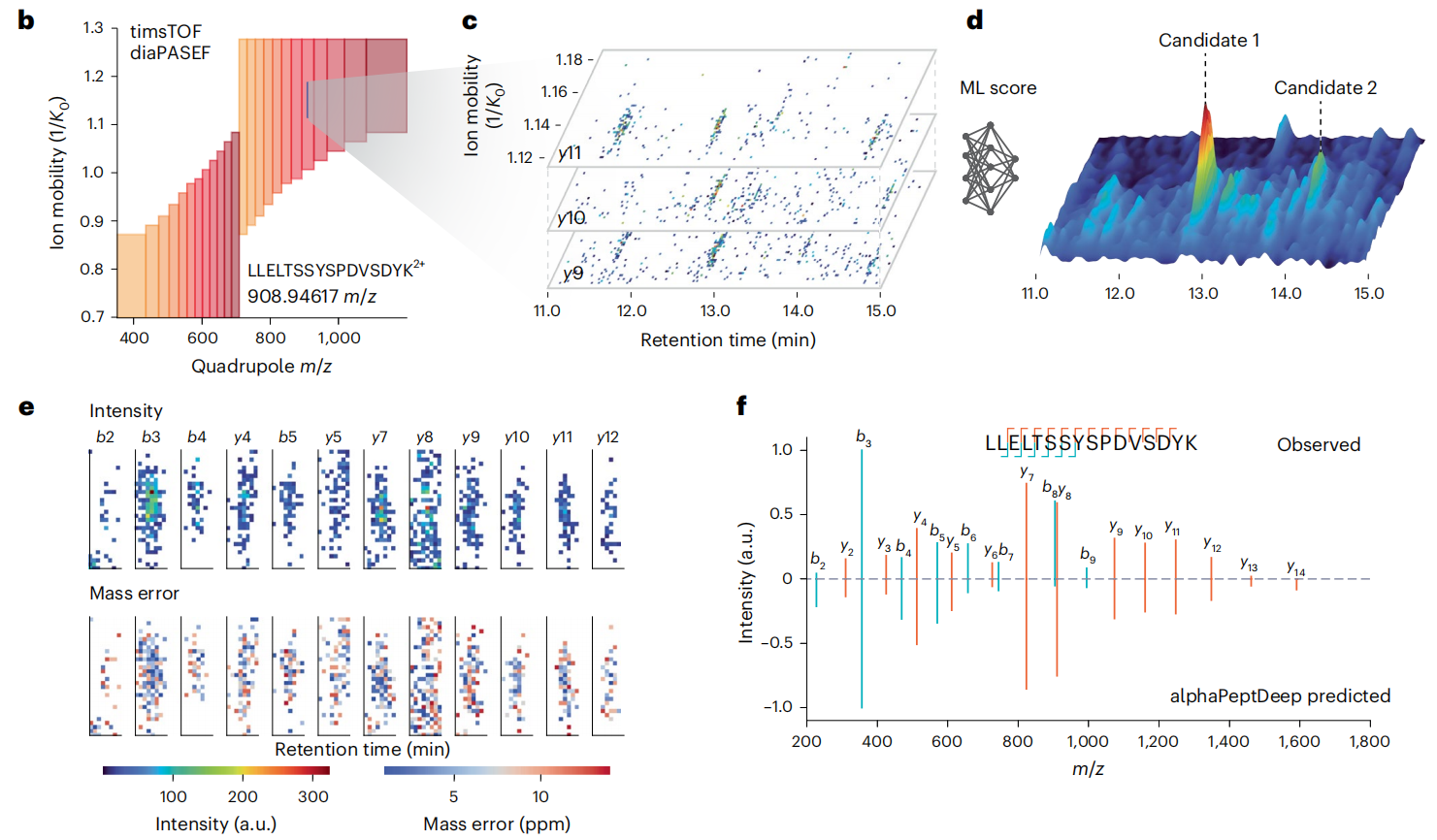
Figure 1
突破2:深度学习 + 迁移学习,不用经验谱库也能精准分析
① 鉴定准:校准+ 竞争,让假阳性 “无处遁形”
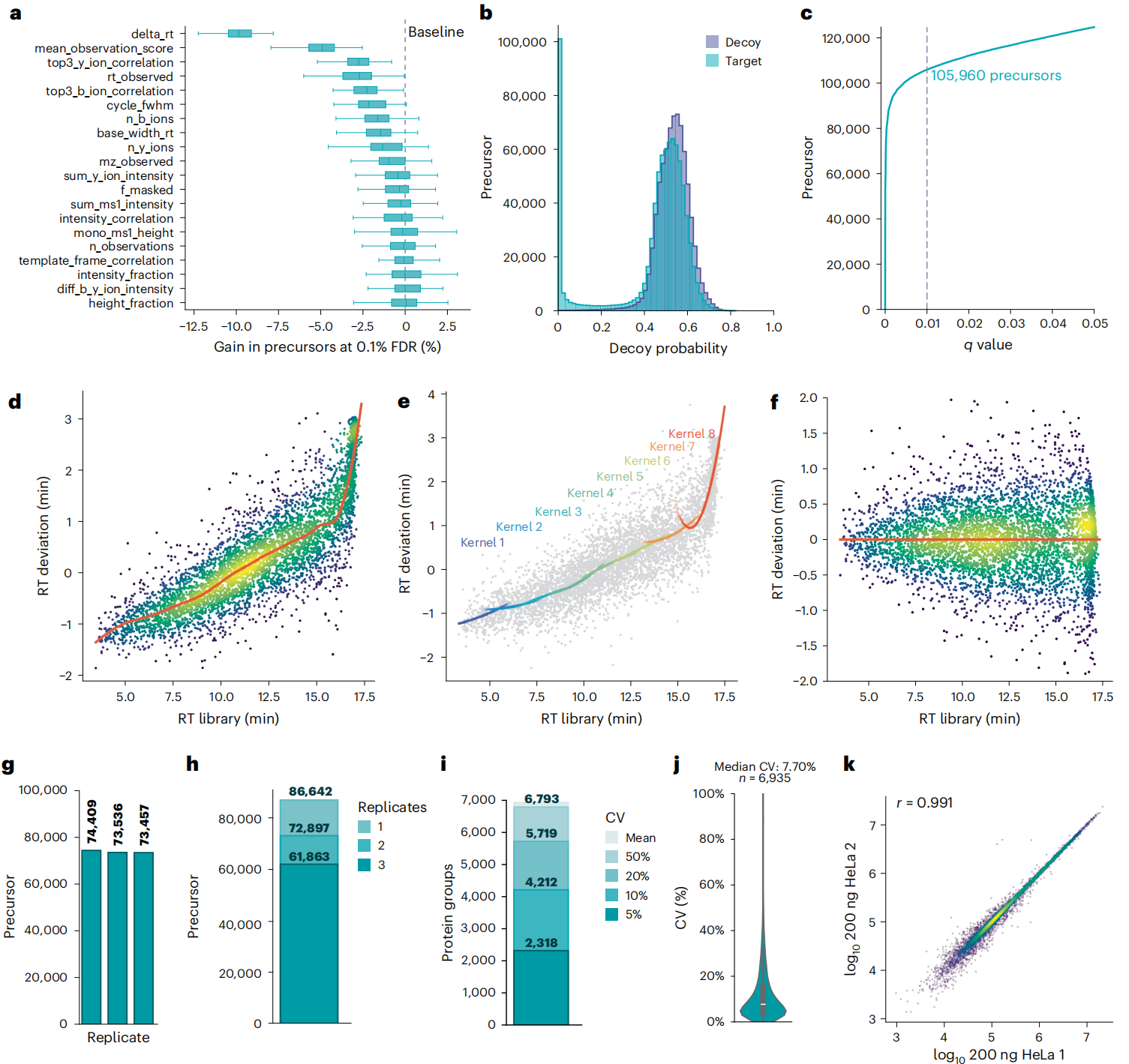
Figure 2
AlphaDIA用全连接神经网络给肽段 “打分”,还会通过算法修正仪器的系统偏差。同时,它还能确保 “一个碎片只对应一个肽段”,避免重复计数,鉴定精度直接拉满。
②不用建库:靠预测谱库搞定分析
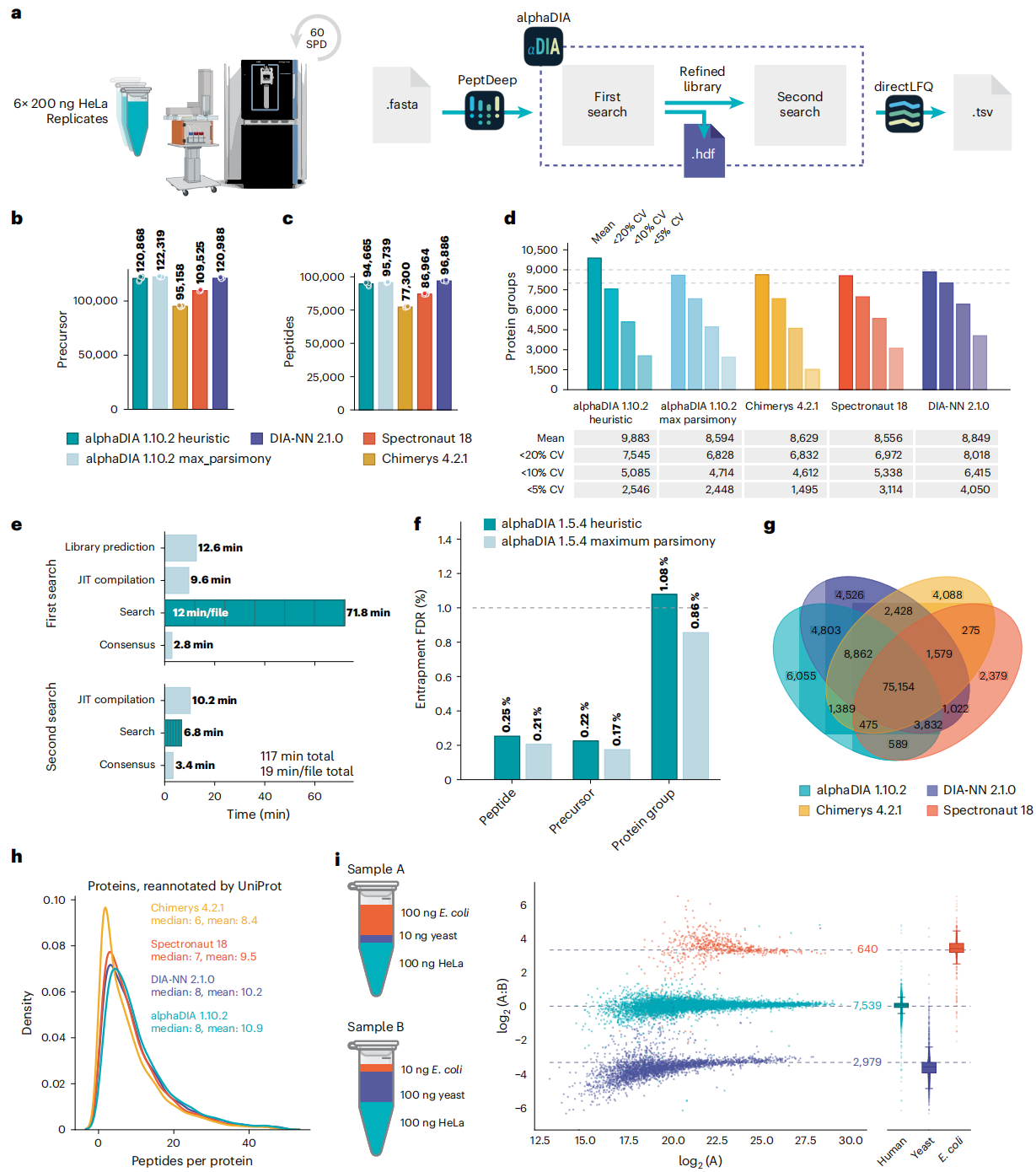
Figure 5
以前做DIA得建“经验谱库”,AlphaDIA 结合团队之前开发的AlphaPeptDeep(一个肽段属性预测工具),能直接构建“预测谱库”,省时又通用。
③适配性强:迁移学习搞定 “未知修饰”
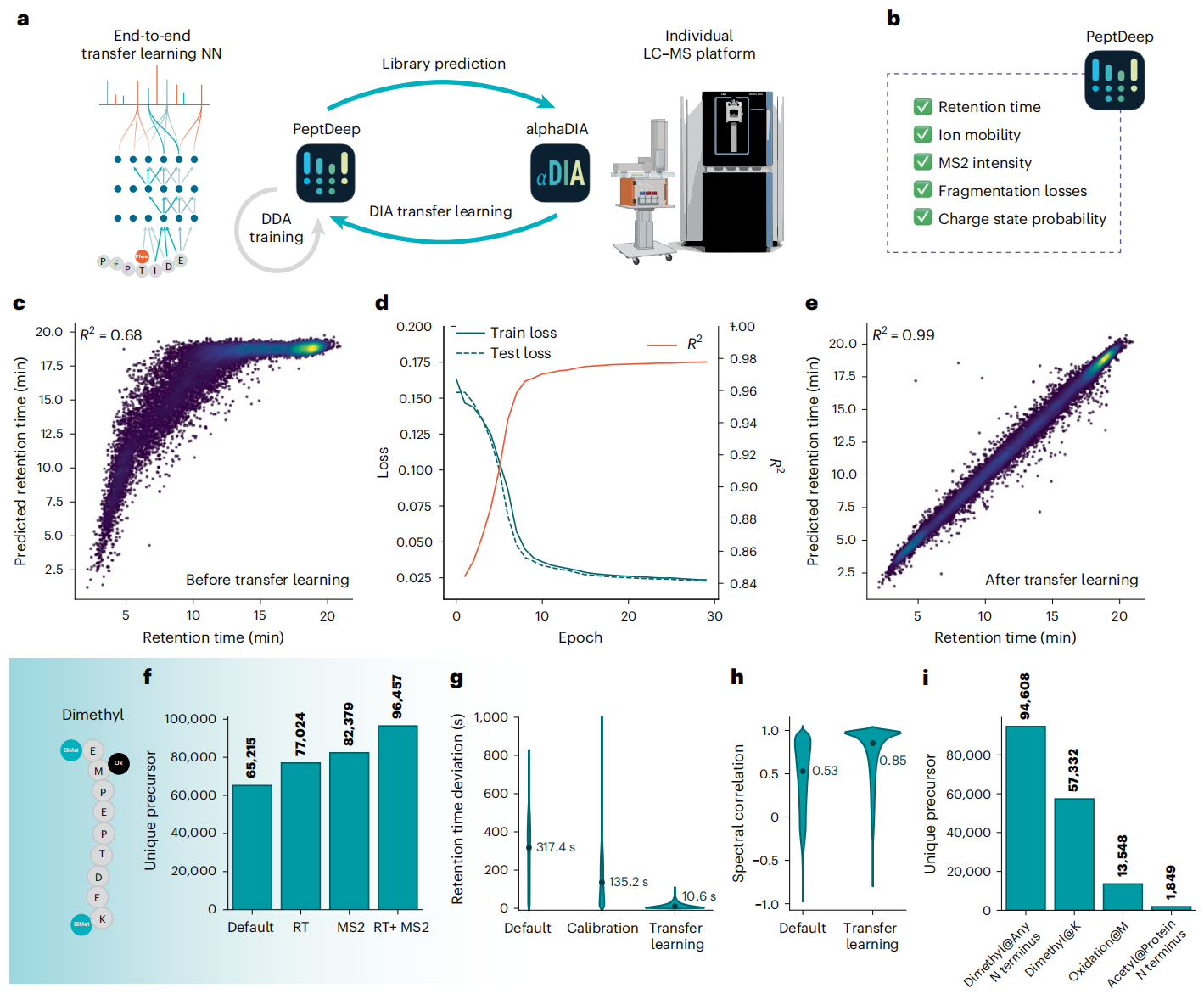
Figure 6
蛋白质的翻译后修饰(PTM)是研究难点,因为不同修饰会改变肽段的“行为”。AlphaDIA的“DIA 迁移学习”能让算法“自己适配”:用初始分析结果训练模型,让模型学会仪器和实验的 “专属特性”,哪怕是没见过的 PTM,也能精准解析。
突破3:跨平台 “通吃”,主流仪器都能搭

Figure 3
三、性能有多强?比主流工具更能打
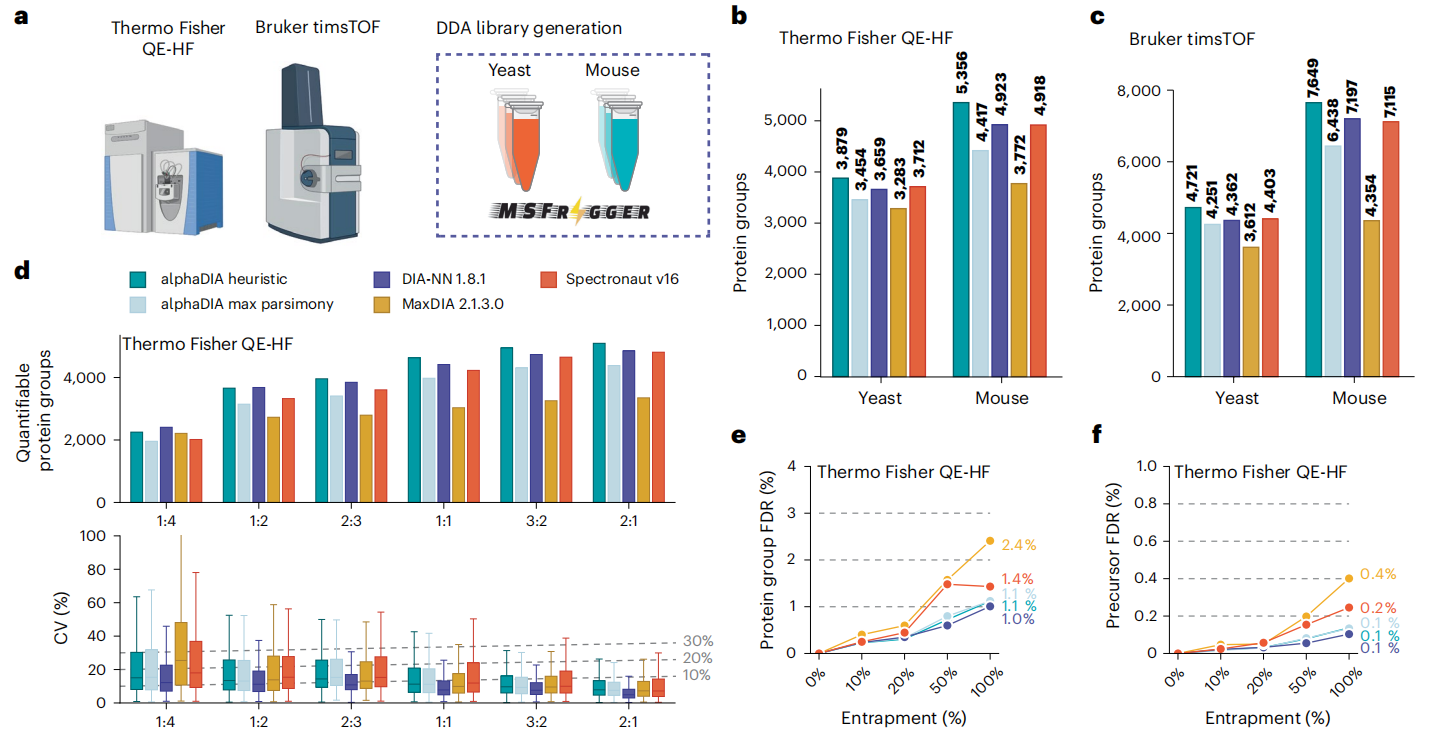
Figure 4
原文链接:https://doi.org/10.1038/s41587-025-02791-w
在生命科学研究里,“蛋白质组学”就像解读生命密码的钥匙——它研究蛋白质及其翻译过程、亚型组成、翻译后修饰(PTM)和降解机制,揭开疾病发生、细胞活动的底层逻辑。在自下而上的蛋白质组学研究中,解决肽段复杂性是基于质谱(MS)的蛋白质组学数据采集和生物信息学分析的核心问题。相比数据依赖采集(DDA),数据非依赖采集(DIA)采用相对较宽的隔离窗口,无偏倚地采集窗口内的所有肽段,动态范围大大提高,实现了更高深度的蛋白质组鉴定,成为单细胞蛋白质组、大规模样本分析的“主力军”。
DIA虽好,却一直有三座“大山”让人头疼
♦ 数据复杂难处理:新一代TOF检测器能捕捉到 “单分子级” 信号,但原始数据里全是无清晰峰形的 “杂乱信号”,传统方法会弄丢关键信息;
♦ 建库太麻烦:以前做DIA得先做DDA预实验,建 “样本专属经验谱库”,耗时耗力还没法通用;
♦ 工具不灵活:主流分析工具多是闭源的,遇到新型仪器(如timsTOF)或未知蛋白质修饰(PTM),直接 “水土不服”。
2025年10月21日,针对DIA分析的“老大难”,Matthias Mann团队在《Nature Biotechnology》发表了题为“AlphaDIA enables DIA transfer learning for feature-free proteomics”的文章,推出开源的AlphaDIA的搜库软件,用“无特征处理 + 深度学习迁移学习”的方式搬平DIA数据解析的三座大山。
一、AlphaDIA的工作流程
Figure 1
AlphaDIA是一款用于DIA搜索的模块化开源框架。该框架基于科学Python工具栈和 alphaPept生态系统构建,支持灵活的搜索策略,可通过Python API、Jupyter notebooks、命令行界面或易于安装的图形用户界面访问。AlphaDIA涵盖了从原始文件到蛋白定量报告的完整工作流程,能够处理所有主流厂商的文件及专有格式。其设计初衷是实现大型队列的“一站式处理”,可在Windows、Linux和Mac系统上原生运行,也能通过Slurm 或Docker以分布式方式在云端部署。
二、AlphaDIA的三大核心突破,每一个都很能打
突破1:“无特征处理”,让TOF数据不再“浪费”
Figure 1
突破2:深度学习 + 迁移学习,不用经验谱库也能精准分析
① 鉴定准:校准+ 竞争,让假阳性 “无处遁形”
Figure 2
AlphaDIA用全连接神经网络给肽段 “打分”,还会通过算法修正仪器的系统偏差。同时,它还能确保 “一个碎片只对应一个肽段”,避免重复计数,鉴定精度直接拉满。
②不用建库:靠预测谱库搞定分析
Figure 5
以前做DIA得建“经验谱库”,AlphaDIA 结合团队之前开发的AlphaPeptDeep(一个肽段属性预测工具),能直接构建“预测谱库”,省时又通用。
③适配性强:迁移学习搞定 “未知修饰”
Figure 6
蛋白质的翻译后修饰(PTM)是研究难点,因为不同修饰会改变肽段的“行为”。AlphaDIA的“DIA 迁移学习”能让算法“自己适配”:用初始分析结果训练模型,让模型学会仪器和实验的 “专属特性”,哪怕是没见过的 PTM,也能精准解析。
突破3:跨平台 “通吃”,主流仪器都能搭
Figure 3
三、性能有多强?比主流工具更能打
Figure 4
原文链接:https://doi.org/10.1038/s41587-025-02791-w


