研究背景
细胞表面蛋白是关键疾病标志物和治疗靶点,但传统核酸适体筛选方法存在通量低、易破坏蛋白天然构象,常遗漏低丰度靶点,且难以同时获取动力学信息等局限,阻碍核酸适体在诊断和治疗中的应用。
2026年1月1日,中国科学院杭州医学研究所/湖南大学谭蔚泓院士与吴芩研究员团队在国际顶尖学术期刊Science发表了题为“SPARK-seq: A high-throughput platform for aptamer discovery and kinetic profiling”的突破性研究成果(第一作者为湖南大学博士生罗国焰,上海交通大学宋佳副研究员为论文共同第一作者)。该研究创新性地将CRISPR基因扰动、单细胞多组学测序(同步捕获mRNA与核酸适体)与深度学习算法相融合,成功构建了名为SPARK-seq的高通量筛选平台,一举实现了对细胞膜表面靶点的大规模发现及高特异性核酸探针(核酸适体)的并行筛选与动力学解析。西湖欧米为本研究中的蛋白质组学(Astral DIA 24-min)检测分析提供了技术支持。
文章标题

西湖欧米为本研究中的蛋白质组学(Astral DIA 24-min)检测分析提供了技术支持
研究结果
01. 全流程整合:SPARK-seq的工作逻辑
研究首先表明,SPARK-seq通过整合CRISPR基因扰动、单细胞多组学测序与Cell-SELEX,构建了完整高通量核酸适体发现流程。其设计的含46-nt随机区的DNA文库经4轮富集后,对目标细胞的结合活性逐步提升(Fig. 1B),可通过特定捕获序列同步检测gRNA、mRNA与核酸适体(Fig. 1C)。后续的计算分析流程能有效整合这三类数据(Fig. 1D),从而将特定的基因扰动事件与对应的核酸适体结合谱直接关联(Fig. 1E),为精准绘制互作网络奠定基础。
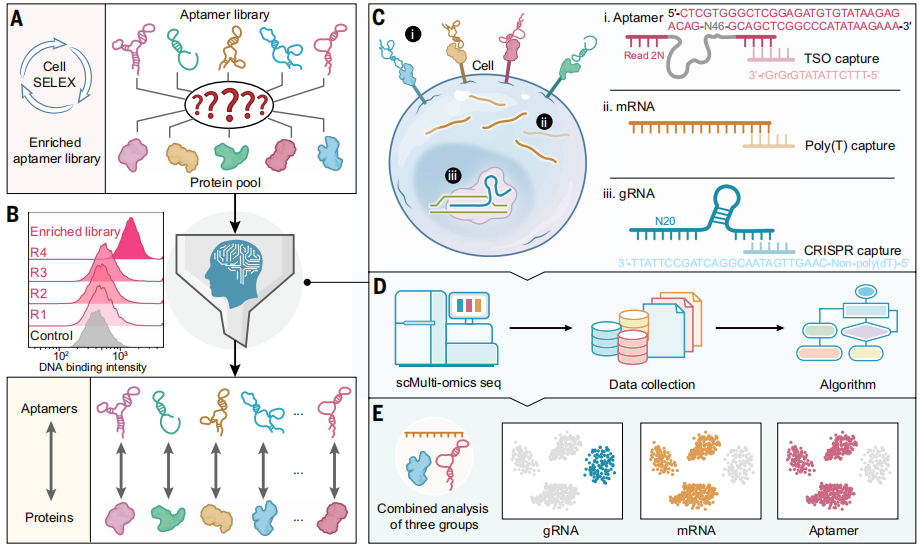
Fig.1 SPARK-seq工作流程总览
02. 概念验证:单细胞层面的精准闭环
为验证平台有效性,研究选取了经前期蛋白质组学分析筛选出的高价值靶点PTK7为对象开展概念验证。Western blot证实,在PTK7基因敲除的细胞中,其蛋白表达显著降低(Fig. 2A)。流式细胞术显示PTK7的特异性核酸适体sgc8c的结合也随之大幅下降(Fig. 2B)。更重要的是,研究将sgc8c改造为平台兼容的验证探针sgc8c-S。单细胞测序结果显示,该探针在PTK7基因敲除细胞中的结合信号显著丢失(Fig. 2C-G),从而在单细胞层面完成了从“基因敲除”到“结合表型丧失”的因果闭环验证。
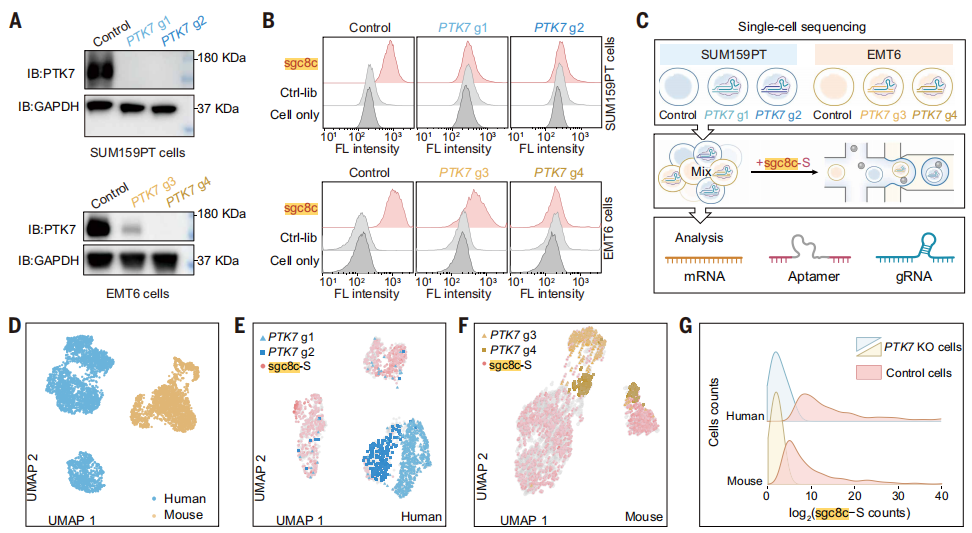
Fig.2 SPARK-seq概念验证实验
03. 高通量映射:从海量数据中锁定互作关系
基于前期蛋白质组学(检测结果覆盖10,970种蛋白质)筛选的13种核心靶点,SPARK-seq展现了强大的高通量发现能力。研究将测序获得的高丰度核酸适体序列聚类成家族(Fig. 3A-B),并通过算法量化它们在各种基因敲除细胞中的结合差异(Fig. 3D)。利用专门开发的SPARTA分析流程(Fig. 3E),最终成功预测并绘制出12个核酸适体家族与9个细胞表面靶蛋白之间的特异性互作网络(Fig. 3F),且结合差异具有高度统计学显著性(Fig. 3G),为后续验证提供高置信度候选对。
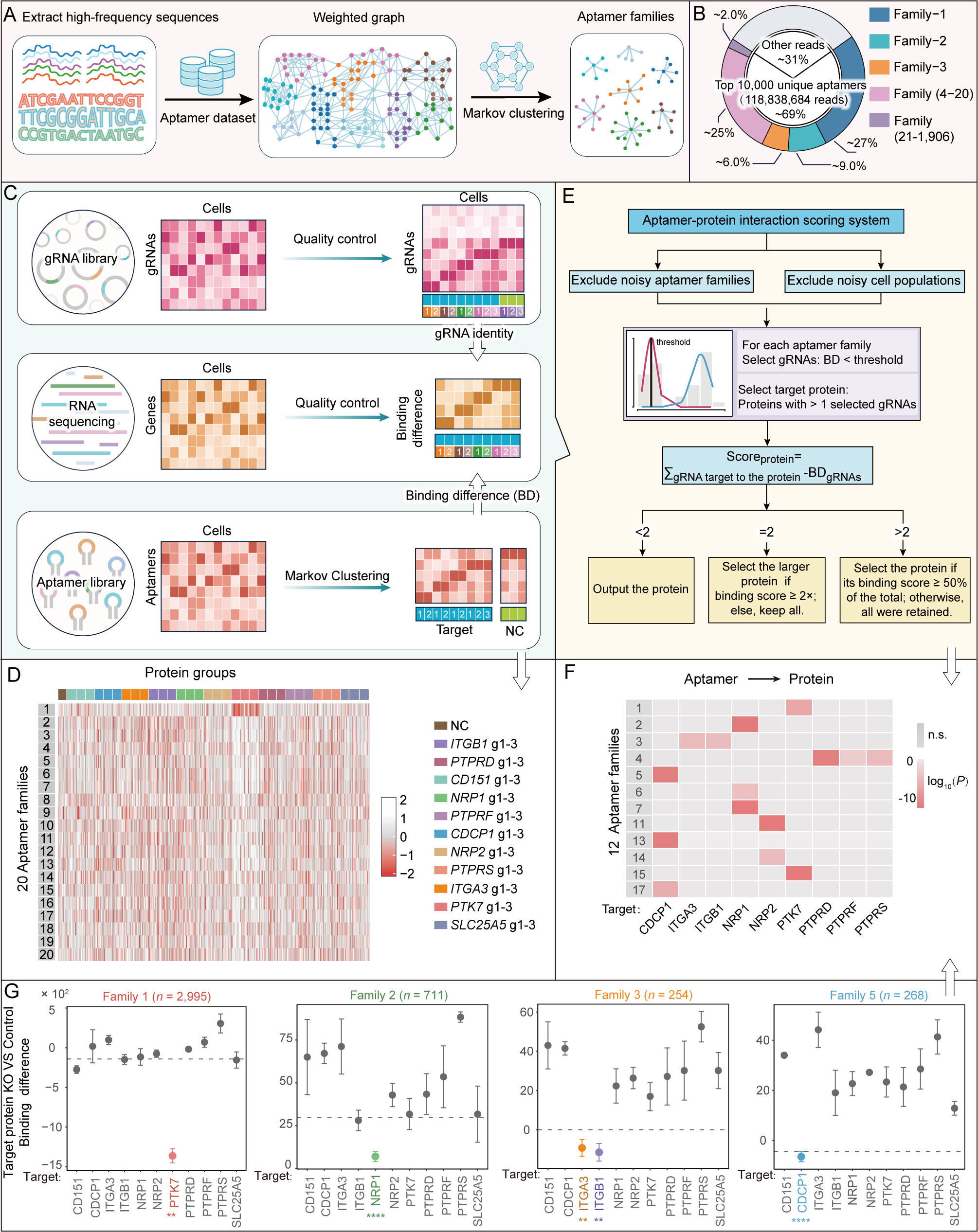
Fig.3 高通量核酸适体-靶点映射流程
04. 正交验证:确认互作关系的可靠性
针对预测的候选对,通过多技术开展系统性验证。例如,流式细胞术证实候选核酸适体仅能结合野生型细胞,而在靶基因敲除细胞上结合消失(Fig. 4B, E);表面等离子共振等技术则验证了其结合亲和力达到纳摩尔水平(Fig. 4C, F)。最终,桑基图清晰地汇总了5535个独特核酸适体序列与8个已验证靶蛋白的精准对应关系(Fig. 4Z),包括能够识别多蛋白复合物或同源蛋白家族的核酸适体,全面证实了映射结果的准确性。
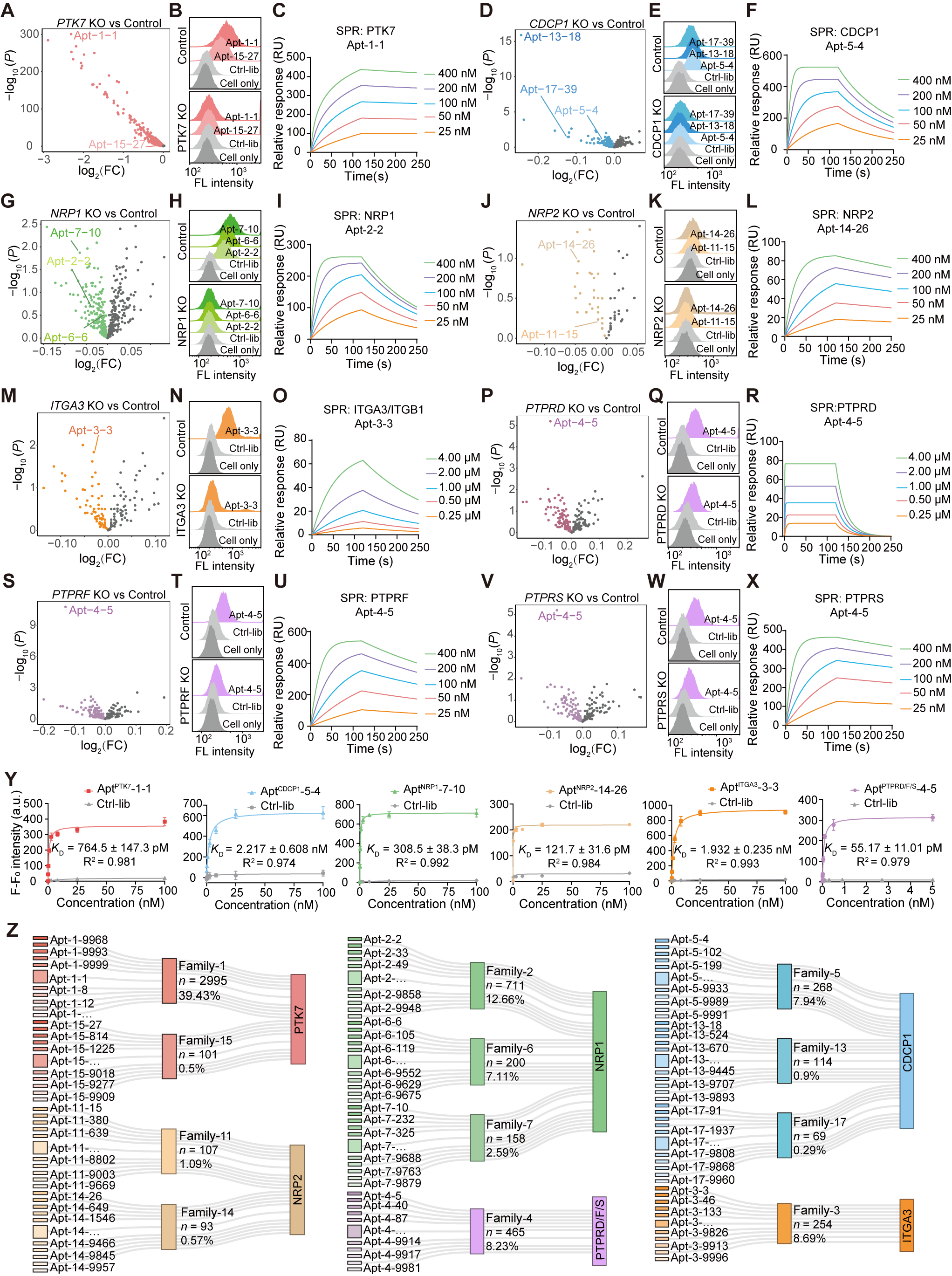
Fig.4 确认核酸适体-靶点结合可靠性
04. 正交验证:确认互作关系的可靠性
针对预测的候选对,通过多技术开展系统性验证。例如,流式细胞术证实候选核酸适体仅能结合野生型细胞,而在靶基因敲除细胞上结合消失(Fig. 4B, E);表面等离子共振等技术则验证了其结合亲和力达到纳摩尔水平(Fig. 4C, F)。最终,桑基图清晰地汇总了5535个独特核酸适体序列与8个已验证靶蛋白的精准对应关系(Fig. 4Z),包括能够识别多蛋白复合物或同源蛋白家族的核酸适体,全面证实了映射结果的准确性。
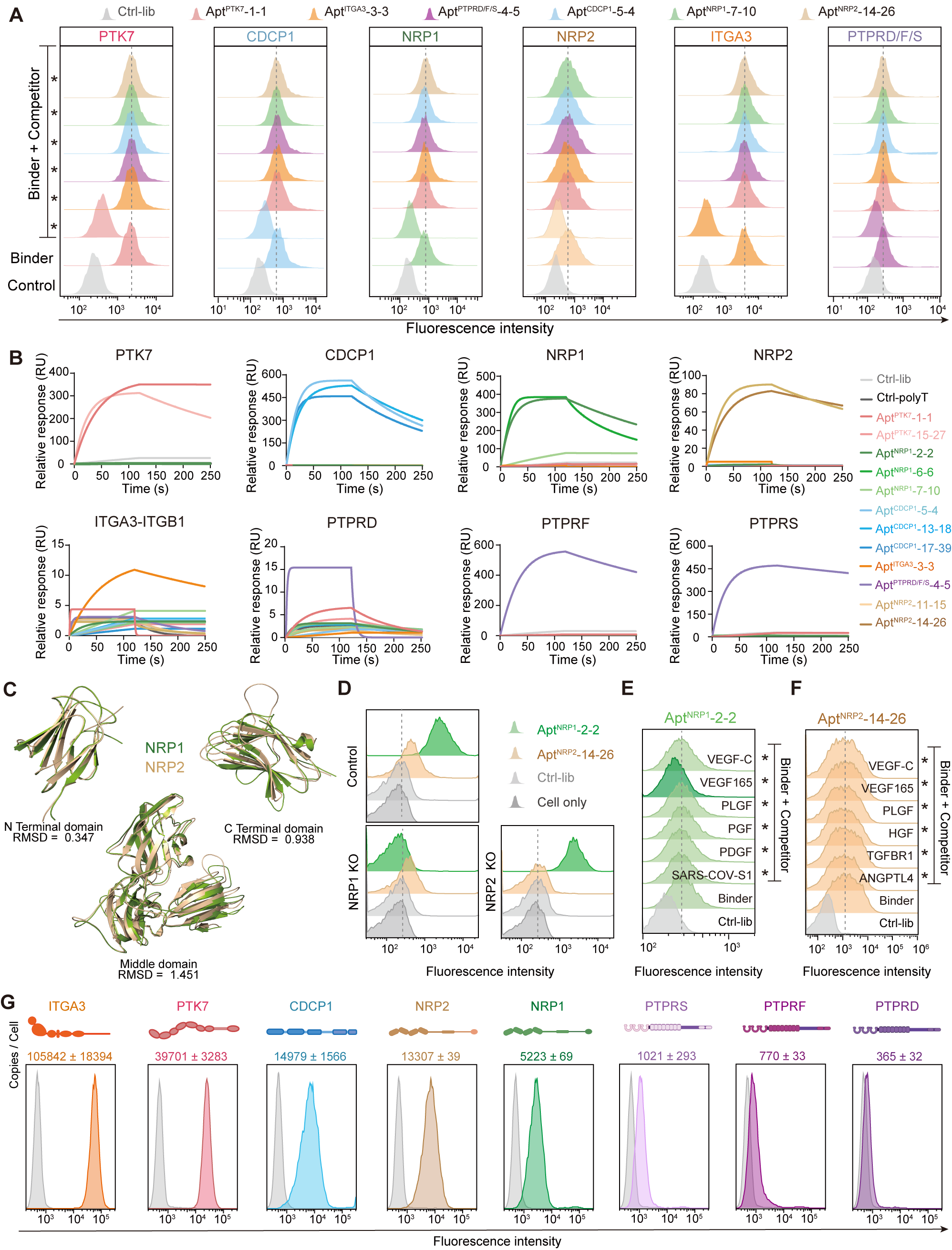
Fig.5 核酸适体高特异性验证
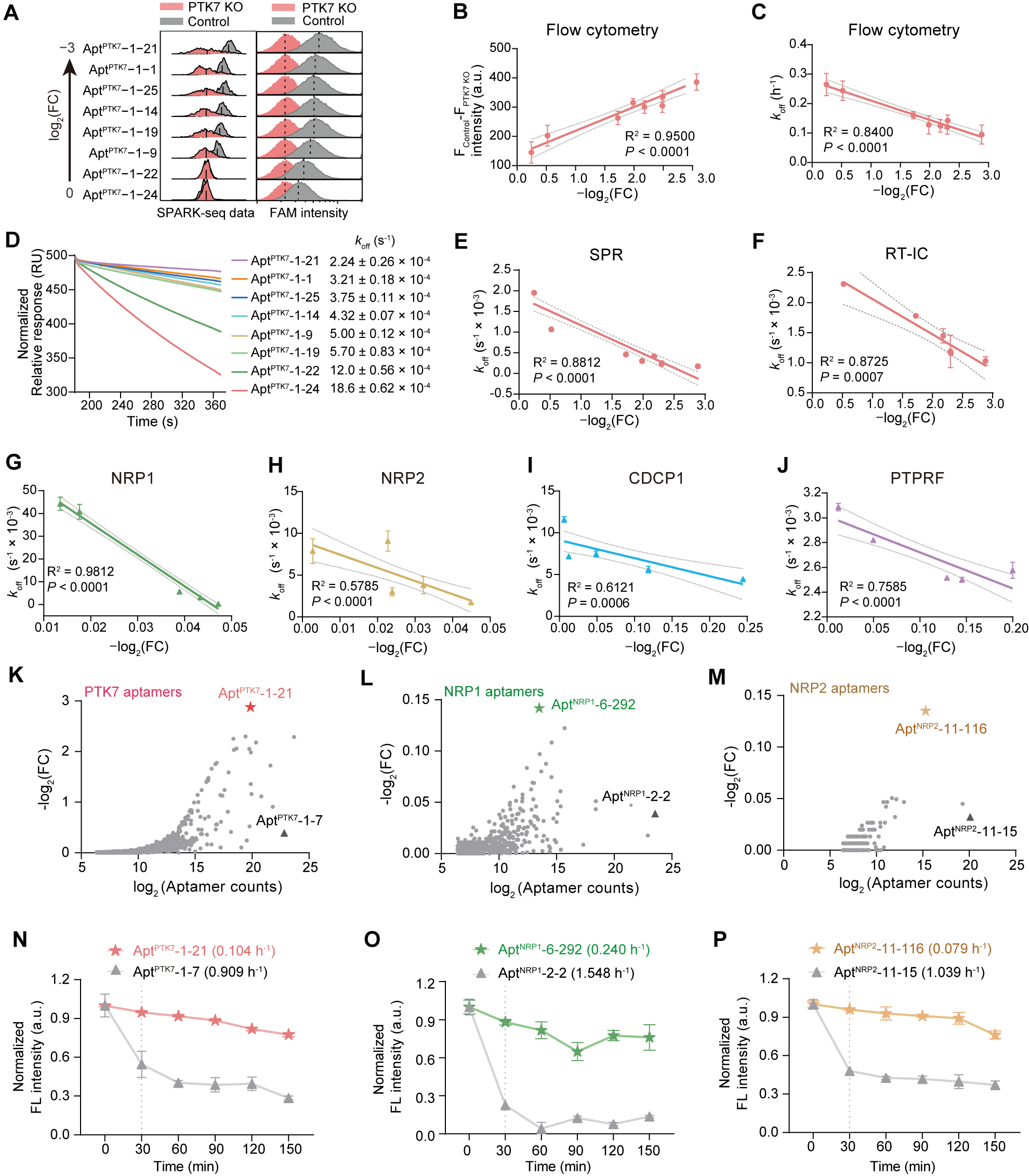
Fig.6 高效筛选慢解离核酸适体
总结与展望
SPARK-seq的核心突破是整合CRISPR基因扰动、单细胞多组学与深度学习算法,成功建立了核酸适体“发现-鉴定-优化”的高通量闭环体系。其中,前期基于Astral DIA 24-min技术的蛋白质组学分析,为锁定关键靶点、引导高效实验设计提供了数据支撑。该平台不仅攻克了传统筛选的通量与信息瓶颈,其产出核酸适体所具备的高特异性与慢解离动力学优势,更为未来开发新一代高灵敏度诊断探针和长效靶向疗法奠定了强大的技术基础,有望加速相关领域从基础研究到临床应用的转化进程。
原文链接:https://www.science.org/doi/10.1126/science.adv6127
研究背景
细胞表面蛋白是关键疾病标志物和治疗靶点,但传统核酸适体筛选方法存在通量低、易破坏蛋白天然构象,常遗漏低丰度靶点,且难以同时获取动力学信息等局限,阻碍核酸适体在诊断和治疗中的应用。
2026年1月1日,中国科学院杭州医学研究所/湖南大学谭蔚泓院士与吴芩研究员团队在国际顶尖学术期刊Science发表了题为“SPARK-seq: A high-throughput platform for aptamer discovery and kinetic profiling”的突破性研究成果(第一作者为湖南大学博士生罗国焰,上海交通大学宋佳副研究员为论文共同第一作者)。该研究创新性地将CRISPR基因扰动、单细胞多组学测序(同步捕获mRNA与核酸适体)与深度学习算法相融合,成功构建了名为SPARK-seq的高通量筛选平台,一举实现了对细胞膜表面靶点的大规模发现及高特异性核酸探针(核酸适体)的并行筛选与动力学解析。西湖欧米为本研究中的蛋白质组学(Astral DIA 24-min)检测分析提供了技术支持。
文章标题
西湖欧米为本研究中的蛋白质组学(Astral DIA 24-min)检测分析提供了技术支持
研究结果
01. 全流程整合:SPARK-seq的工作逻辑
研究首先表明,SPARK-seq通过整合CRISPR基因扰动、单细胞多组学测序与Cell-SELEX,构建了完整高通量核酸适体发现流程。其设计的含46-nt随机区的DNA文库经4轮富集后,对目标细胞的结合活性逐步提升(Fig. 1B),可通过特定捕获序列同步检测gRNA、mRNA与核酸适体(Fig. 1C)。后续的计算分析流程能有效整合这三类数据(Fig. 1D),从而将特定的基因扰动事件与对应的核酸适体结合谱直接关联(Fig. 1E),为精准绘制互作网络奠定基础。
Fig.1 SPARK-seq工作流程总览
02. 概念验证:单细胞层面的精准闭环
为验证平台有效性,研究选取了经前期蛋白质组学分析筛选出的高价值靶点PTK7为对象开展概念验证。Western blot证实,在PTK7基因敲除的细胞中,其蛋白表达显著降低(Fig. 2A)。流式细胞术显示PTK7的特异性核酸适体sgc8c的结合也随之大幅下降(Fig. 2B)。更重要的是,研究将sgc8c改造为平台兼容的验证探针sgc8c-S。单细胞测序结果显示,该探针在PTK7基因敲除细胞中的结合信号显著丢失(Fig. 2C-G),从而在单细胞层面完成了从“基因敲除”到“结合表型丧失”的因果闭环验证。
Fig.2 SPARK-seq概念验证实验
03. 高通量映射:从海量数据中锁定互作关系
基于前期蛋白质组学(检测结果覆盖10,970种蛋白质)筛选的13种核心靶点,SPARK-seq展现了强大的高通量发现能力。研究将测序获得的高丰度核酸适体序列聚类成家族(Fig. 3A-B),并通过算法量化它们在各种基因敲除细胞中的结合差异(Fig. 3D)。利用专门开发的SPARTA分析流程(Fig. 3E),最终成功预测并绘制出12个核酸适体家族与9个细胞表面靶蛋白之间的特异性互作网络(Fig. 3F),且结合差异具有高度统计学显著性(Fig. 3G),为后续验证提供高置信度候选对。
Fig.3 高通量核酸适体-靶点映射流程
04. 正交验证:确认互作关系的可靠性
针对预测的候选对,通过多技术开展系统性验证。例如,流式细胞术证实候选核酸适体仅能结合野生型细胞,而在靶基因敲除细胞上结合消失(Fig. 4B, E);表面等离子共振等技术则验证了其结合亲和力达到纳摩尔水平(Fig. 4C, F)。最终,桑基图清晰地汇总了5535个独特核酸适体序列与8个已验证靶蛋白的精准对应关系(Fig. 4Z),包括能够识别多蛋白复合物或同源蛋白家族的核酸适体,全面证实了映射结果的准确性。
Fig.4 确认核酸适体-靶点结合可靠性
04. 正交验证:确认互作关系的可靠性
针对预测的候选对,通过多技术开展系统性验证。例如,流式细胞术证实候选核酸适体仅能结合野生型细胞,而在靶基因敲除细胞上结合消失(Fig. 4B, E);表面等离子共振等技术则验证了其结合亲和力达到纳摩尔水平(Fig. 4C, F)。最终,桑基图清晰地汇总了5535个独特核酸适体序列与8个已验证靶蛋白的精准对应关系(Fig. 4Z),包括能够识别多蛋白复合物或同源蛋白家族的核酸适体,全面证实了映射结果的准确性。
Fig.5 核酸适体高特异性验证
Fig.6 高效筛选慢解离核酸适体
总结与展望
SPARK-seq的核心突破是整合CRISPR基因扰动、单细胞多组学与深度学习算法,成功建立了核酸适体“发现-鉴定-优化”的高通量闭环体系。其中,前期基于Astral DIA 24-min技术的蛋白质组学分析,为锁定关键靶点、引导高效实验设计提供了数据支撑。该平台不仅攻克了传统筛选的通量与信息瓶颈,其产出核酸适体所具备的高特异性与慢解离动力学优势,更为未来开发新一代高灵敏度诊断探针和长效靶向疗法奠定了强大的技术基础,有望加速相关领域从基础研究到临床应用的转化进程。
原文链接:https://www.science.org/doi/10.1126/science.adv6127