1 (Cell Discov) 结直肠癌免疫微空间:巨噬细胞-T细胞互作驱动免疫治疗响应
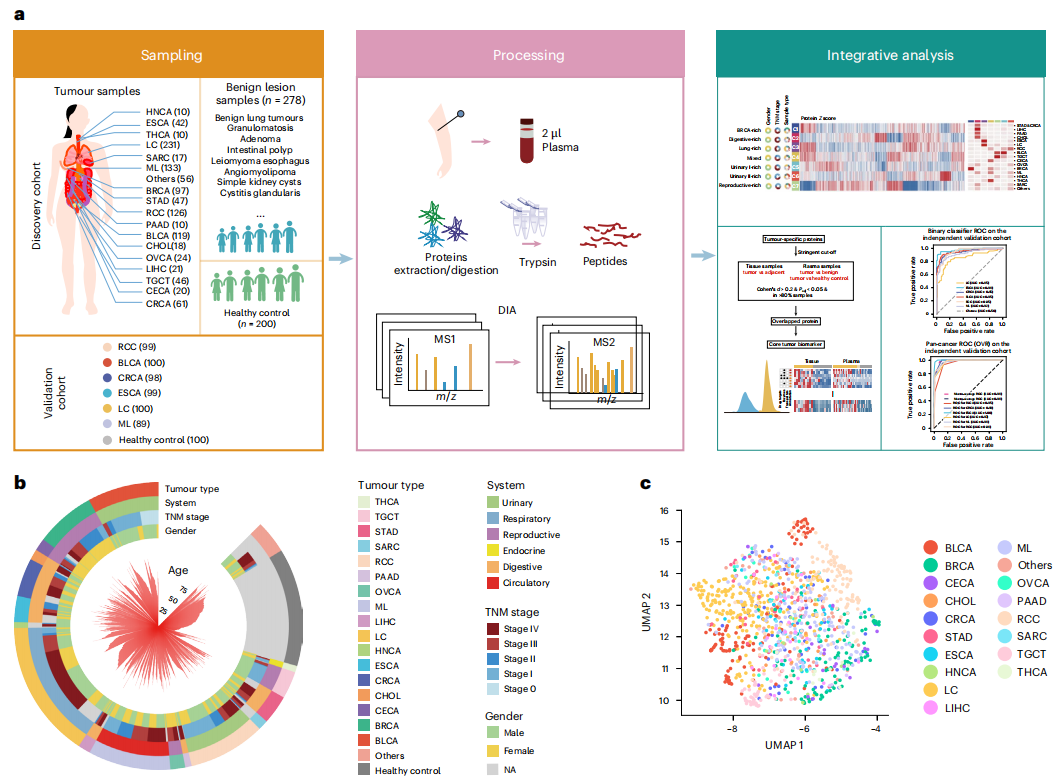
2 (Nature Biomed Eng) 泛癌血浆蛋白质组图谱揭示代谢-免疫交互新机制
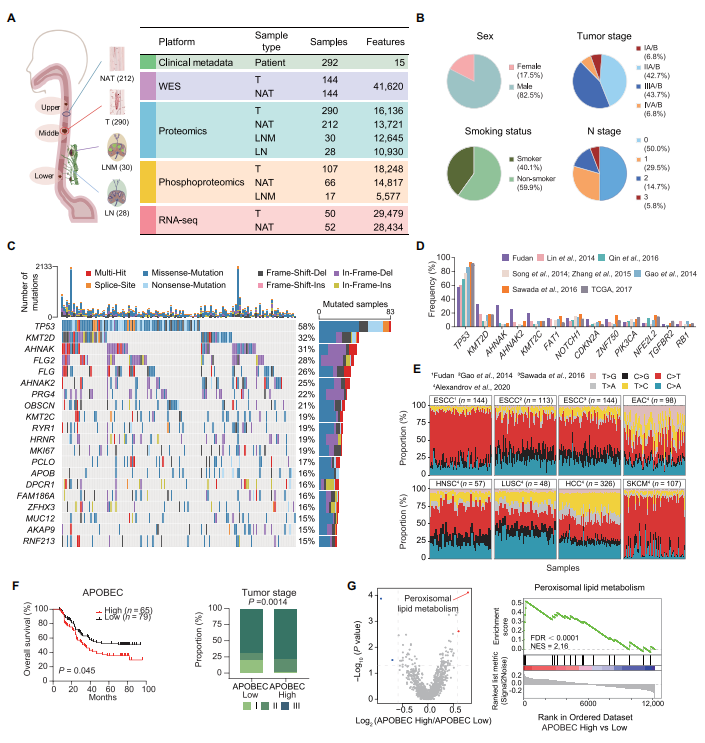
3 (Sci Transl Med) 食管鳞癌蛋白-基因组学解析染色体扩增驱动机制
4 (Mol Cell Proteomics) 鸟枪法蛋白质组学实现广谱病毒精准筛查
5 (iScience) 结构互作组学预测遗传病分子机制
6 (iMeta) 宏蛋白质组学技术指南推动微生物组功能解析
1.(Cell Discovery,IF:38.1)结直肠癌免疫微空间:巨噬细胞-T细胞互作驱动免疫治疗响应

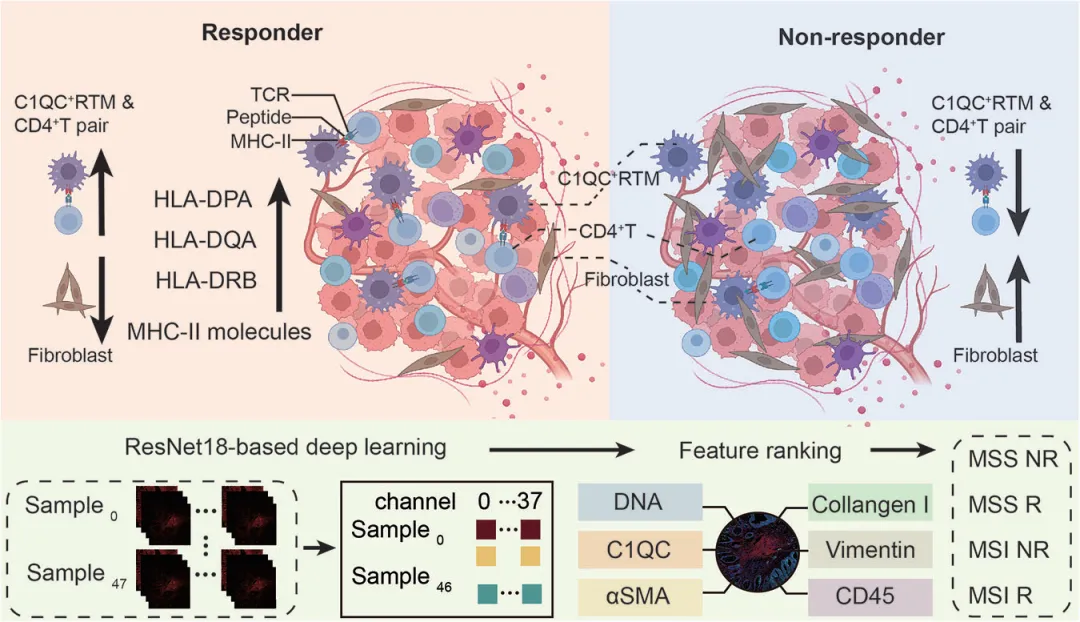
2.(Nature Biomed Eng,IF:27.7)泛癌血浆蛋白质组图谱揭示代谢-免疫交互新机制

3.(Sci Transl Med,IF:17.1)食管鳞癌蛋白-基因组学解析染色体扩增驱动机制

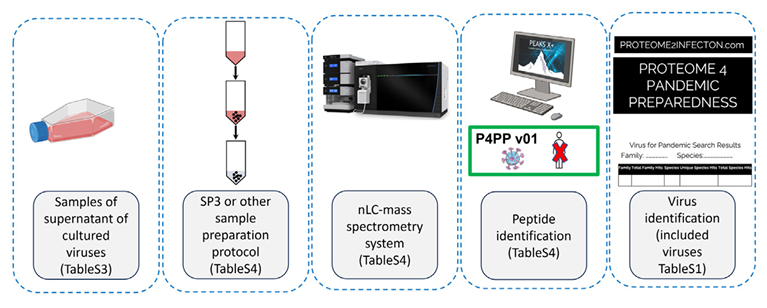
4.(Mol Cell Proteomics,IF:6.1)鸟枪法蛋白质组学实现广谱病毒精准筛查

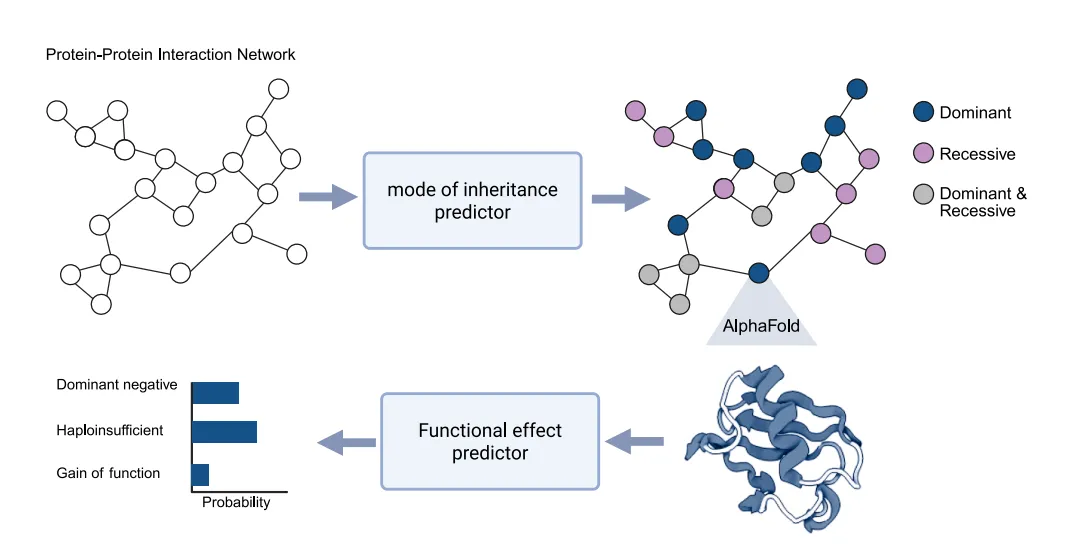
5.(iScience,IF:4.6)结构互作组学预测遗传病分子机制

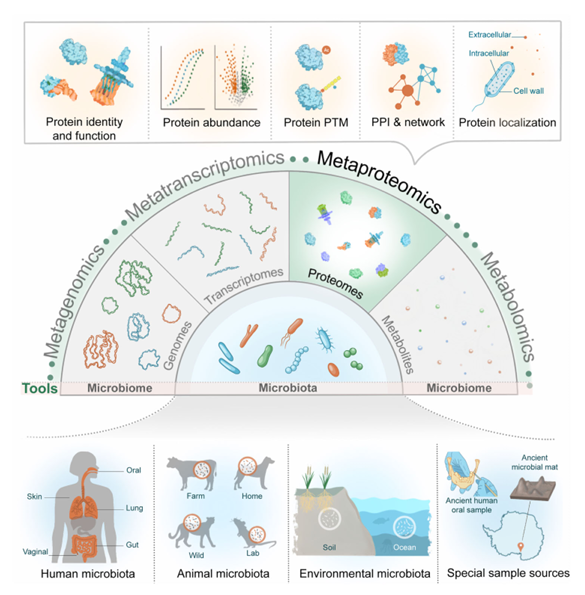
6.(iMeta,IF:23.8)宏蛋白质组学技术指南推动微生物组功能解析

免责声明:本篇文章由人工智能(ChatGPT 4o)撰写,内容基于相关文献、研究成果和现有科技进展的综合分析。虽然我们力求确保文章信息的准确性和可靠性,但由于AI生成内容的局限性,本文的观点和见解仅供参考。读者在应用或引用本文内容时,请自行核实相关信息和数据的有效性。我们不对任何因使用本文内容所导致的直接或间接损失承担责任。
1 (Cell Discov) 结直肠癌免疫微空间:巨噬细胞-T细胞互作驱动免疫治疗响应
2 (Nature Biomed Eng) 泛癌血浆蛋白质组图谱揭示代谢-免疫交互新机制
3 (Sci Transl Med) 食管鳞癌蛋白-基因组学解析染色体扩增驱动机制
4 (Mol Cell Proteomics) 鸟枪法蛋白质组学实现广谱病毒精准筛查
5 (iScience) 结构互作组学预测遗传病分子机制
6 (iMeta) 宏蛋白质组学技术指南推动微生物组功能解析
1.(Cell Discovery,IF:38.1)结直肠癌免疫微空间:巨噬细胞-T细胞互作驱动免疫治疗响应
2.(Nature Biomed Eng,IF:27.7)泛癌血浆蛋白质组图谱揭示代谢-免疫交互新机制
3.(Sci Transl Med,IF:17.1)食管鳞癌蛋白-基因组学解析染色体扩增驱动机制
4.(Mol Cell Proteomics,IF:6.1)鸟枪法蛋白质组学实现广谱病毒精准筛查
5.(iScience,IF:4.6)结构互作组学预测遗传病分子机制
6.(iMeta,IF:23.8)宏蛋白质组学技术指南推动微生物组功能解析
免责声明:本篇文章由人工智能(ChatGPT 4o)撰写,内容基于相关文献、研究成果和现有科技进展的综合分析。虽然我们力求确保文章信息的准确性和可靠性,但由于AI生成内容的局限性,本文的观点和见解仅供参考。读者在应用或引用本文内容时,请自行核实相关信息和数据的有效性。我们不对任何因使用本文内容所导致的直接或间接损失承担责任。