






♦ midiaPASEF采样模式
图源:论文截图
Stefan Tenzer团队提出了midiaPASEF (maximizes information) 采样模式,和synchro-PASEF类似采用了四级杆“sliding”方法,吻合淌度轴-质量轴对角线的母离子云形状。不同点主要在于midiaPASEF方法中相邻的MIDIA frames间存在12Th重叠,使得每个碎片离子可在一个cycle time的3/20 MIDIA frames中检测到,overall cycle time的时长也延长到2.2s。
此外他们开发了基于Snakemake的MIDIAID pipeline,对midiaPASEF文件进行自动化的多维反卷积处理,通过机器学习迭代优化母离子-碎片离子匹配结果,生成“人眼几乎无法区分”的DDA样谱图和MGF文件,可以直接被PEASKS、FragPipe等软件分析。使用150 μm x 150 mm column,30 min梯度分离300ng HeLa消化肽,midiaPASEF每分钟最高unique peptides鉴定量是ddaPASEF的1.6倍 (2400/1500)。不过本文章(预印本)中缺少LFQ定量结果的benchmark,其准确性有待进一步验证。
https://doi.org/10.1101/2023.01.30.526204
♦ Deep learning in Proteomics

图源:论文截图
来自贝勒医学院的Bing Zhang团队于2020年在Proteomics发表了综述文章,阐述了当时AI在蛋白质组学中的主要任务:
保留时间预测:根据肽段序列预测其在液相色谱中的分离时间,有助于提高肽段鉴定和定量的效率和准确性;MS/MS谱图预测:根据肽段序列预测其对应的碎片离子谱图;从头测序:直接从MS/MS谱图推断出肽段序列,而不依赖于蛋白质数据库,有助于发现新颖或未知的肽段或蛋白质;翻译后修饰预测:根据肽段序列或MS/MS谱图预测其可能存在的翻译后修饰类型和位置,有助于揭示蛋白质功能调控机制;主要组织相容性复合物-肽结合预测:根据肽段序列或HLA分子类型预测其与HLA分子的结合亲和力或稳定性,有助于研究免疫系统识别抗原的过程;蛋白质结构预测:根据蛋白质序列预测其三级结构或二级结构元素,有助于理解蛋白质功能和相互作用。
https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/pmic.201900335
♦ 从手工到深度特征的图像匹配的概述

图源:论文截图

图源:文章Figure
这篇文献是一篇名为“Image Matching from Handcrafted to Deep Features: A Survey”的论文,发表在2021年的《International Journal of Computer Vision》上。
这篇文章系统地回顾和分析了图像匹配领域中的经典的和最新的技术。在介绍基于特征的图像匹配流程的基础上,本文首先介绍了从手工方法到可训练方法的特征检测、描述和匹配技术,并对这些方法在理论和实践中的发展进行了分析。其次,本文简要介绍了几种典型的基于图像匹配的应用,以便全面理解图像匹配的重要性。此外,本文还通过对代表性数据集的广泛实验,对这些经典和最新的技术进行了全面客观的比较。最后,本文总结了图像匹配技术的现状,并对未来的研究方向进行了深入的探讨和展望。
有一个结论是,在图形匹配方面,FGM和TM的性能比较优秀,但是TM的效率更高。这个结论可能对目前正在进行的甲状腺结节DIAT数据表型预测工作中的DIAT特征区域-肽段匹配任务有所帮助。
♦ 细胞新合成的蛋白质
图源:论文截图
图源:文章Figure
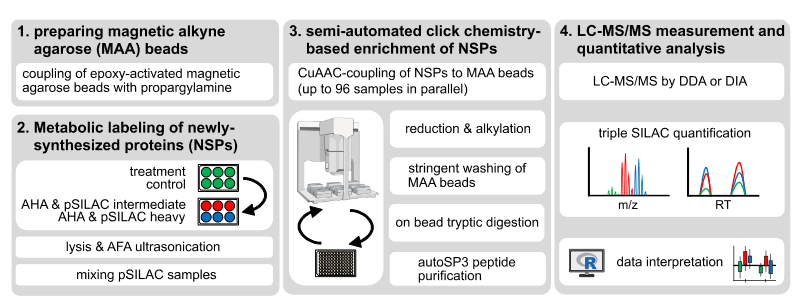
来自德国癌症研究中心DKFZ开发SP3技术的Jeroen Krijgsveld课题组3月3日在bioRxiv发表了一篇针对于细胞新合成的蛋白质(newly synthesized proteins. NSP)的工作。
NSP蛋白一般使用Ribosomal profling的技术进行监测,但是还是依赖于RNA测序后的间接推断、非活性核糖体也无法进行监测在这在药物突变实验中很常见。
因此,这个项目的设计了在质谱蛋白质组层面进行NSP的检测,通过插入嘌呤霉素(或其衍生物)、同位素标记的氨基酸、非天然氨基酸或其组合的进行tripleSILAC和pulse SILAC标记, 也利用了96孔板自动化装置在样本实现autoSP3的优化,并且也在DIA层面结合了plexDIA的数据分析方法来进行蛋白定量,最终他们优化并建立了一个体系化的流程QuanNPA,应用的展示是干扰素-γ (IFNg) 诱导的动态细胞影响,使用了HeLa细胞系来对新靶标检测。最终90min梯度Obitrap plexDIA分析方法鉴定到了8130个蛋白组和超过六千个蛋白保持了18.1%的缺失率,并且找到了时序差异表达的蛋白。
该工作提升了sp3的实验通量也整合了最新的plexDIA数据分析方法并展示了生物学的一个应用是一个值得关注的技术性工作。
♦ midiaPASEF采样模式
图源:论文截图
Stefan Tenzer团队提出了midiaPASEF (maximizes information) 采样模式,和synchro-PASEF类似采用了四级杆“sliding”方法,吻合淌度轴-质量轴对角线的母离子云形状。不同点主要在于midiaPASEF方法中相邻的MIDIA frames间存在12Th重叠,使得每个碎片离子可在一个cycle time的3/20 MIDIA frames中检测到,overall cycle time的时长也延长到2.2s。
此外他们开发了基于Snakemake的MIDIAID pipeline,对midiaPASEF文件进行自动化的多维反卷积处理,通过机器学习迭代优化母离子-碎片离子匹配结果,生成“人眼几乎无法区分”的DDA样谱图和MGF文件,可以直接被PEASKS、FragPipe等软件分析。使用150 μm x 150 mm column,30 min梯度分离300ng HeLa消化肽,midiaPASEF每分钟最高unique peptides鉴定量是ddaPASEF的1.6倍 (2400/1500)。不过本文章(预印本)中缺少LFQ定量结果的benchmark,其准确性有待进一步验证。
https://doi.org/10.1101/2023.01.30.526204
♦ Deep learning in Proteomics
图源:论文截图
来自贝勒医学院的Bing Zhang团队于2020年在Proteomics发表了综述文章,阐述了当时AI在蛋白质组学中的主要任务:
保留时间预测:根据肽段序列预测其在液相色谱中的分离时间,有助于提高肽段鉴定和定量的效率和准确性;MS/MS谱图预测:根据肽段序列预测其对应的碎片离子谱图;从头测序:直接从MS/MS谱图推断出肽段序列,而不依赖于蛋白质数据库,有助于发现新颖或未知的肽段或蛋白质;翻译后修饰预测:根据肽段序列或MS/MS谱图预测其可能存在的翻译后修饰类型和位置,有助于揭示蛋白质功能调控机制;主要组织相容性复合物-肽结合预测:根据肽段序列或HLA分子类型预测其与HLA分子的结合亲和力或稳定性,有助于研究免疫系统识别抗原的过程;蛋白质结构预测:根据蛋白质序列预测其三级结构或二级结构元素,有助于理解蛋白质功能和相互作用。
https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/pmic.201900335
♦ 从手工到深度特征的图像匹配的概述
图源:论文截图
图源:文章Figure
这篇文献是一篇名为“Image Matching from Handcrafted to Deep Features: A Survey”的论文,发表在2021年的《International Journal of Computer Vision》上。
这篇文章系统地回顾和分析了图像匹配领域中的经典的和最新的技术。在介绍基于特征的图像匹配流程的基础上,本文首先介绍了从手工方法到可训练方法的特征检测、描述和匹配技术,并对这些方法在理论和实践中的发展进行了分析。其次,本文简要介绍了几种典型的基于图像匹配的应用,以便全面理解图像匹配的重要性。此外,本文还通过对代表性数据集的广泛实验,对这些经典和最新的技术进行了全面客观的比较。最后,本文总结了图像匹配技术的现状,并对未来的研究方向进行了深入的探讨和展望。
有一个结论是,在图形匹配方面,FGM和TM的性能比较优秀,但是TM的效率更高。这个结论可能对目前正在进行的甲状腺结节DIAT数据表型预测工作中的DIAT特征区域-肽段匹配任务有所帮助。
♦ 细胞新合成的蛋白质
图源:论文截图
图源:文章Figure
来自德国癌症研究中心DKFZ开发SP3技术的Jeroen Krijgsveld课题组3月3日在bioRxiv发表了一篇针对于细胞新合成的蛋白质(newly synthesized proteins. NSP)的工作。
NSP蛋白一般使用Ribosomal profling的技术进行监测,但是还是依赖于RNA测序后的间接推断、非活性核糖体也无法进行监测在这在药物突变实验中很常见。
因此,这个项目的设计了在质谱蛋白质组层面进行NSP的检测,通过插入嘌呤霉素(或其衍生物)、同位素标记的氨基酸、非天然氨基酸或其组合的进行tripleSILAC和pulse SILAC标记, 也利用了96孔板自动化装置在样本实现autoSP3的优化,并且也在DIA层面结合了plexDIA的数据分析方法来进行蛋白定量,最终他们优化并建立了一个体系化的流程QuanNPA,应用的展示是干扰素-γ (IFNg) 诱导的动态细胞影响,使用了HeLa细胞系来对新靶标检测。最终90min梯度Obitrap plexDIA分析方法鉴定到了8130个蛋白组和超过六千个蛋白保持了18.1%的缺失率,并且找到了时序差异表达的蛋白。
该工作提升了sp3的实验通量也整合了最新的plexDIA数据分析方法并展示了生物学的一个应用是一个值得关注的技术性工作。


