随着质谱技术的飞速发展,大规模、高维度数据的大量产生为疾病研究提供了丰富的资源。蛋白质组学大队列研究通过揭示疾病机制、发现生物标志物和识别治疗靶点,显著推动了个性化医疗的发展。
西湖欧米专注于临床大队列解决方案,包括分子分型、预后分析、机器学习、时间序列分析等。机器学习通过利用复杂算法构建模型,高效地从海量数据中提取关键特征,在早期诊断和疾病分期、预后等相关生物标志物研究中得到了广泛应用,可显著提高疾病预测和个体化治疗的精准度,推动精准医学的发展。
PART 1
西湖欧米临床大队列——
机器学习研究思路
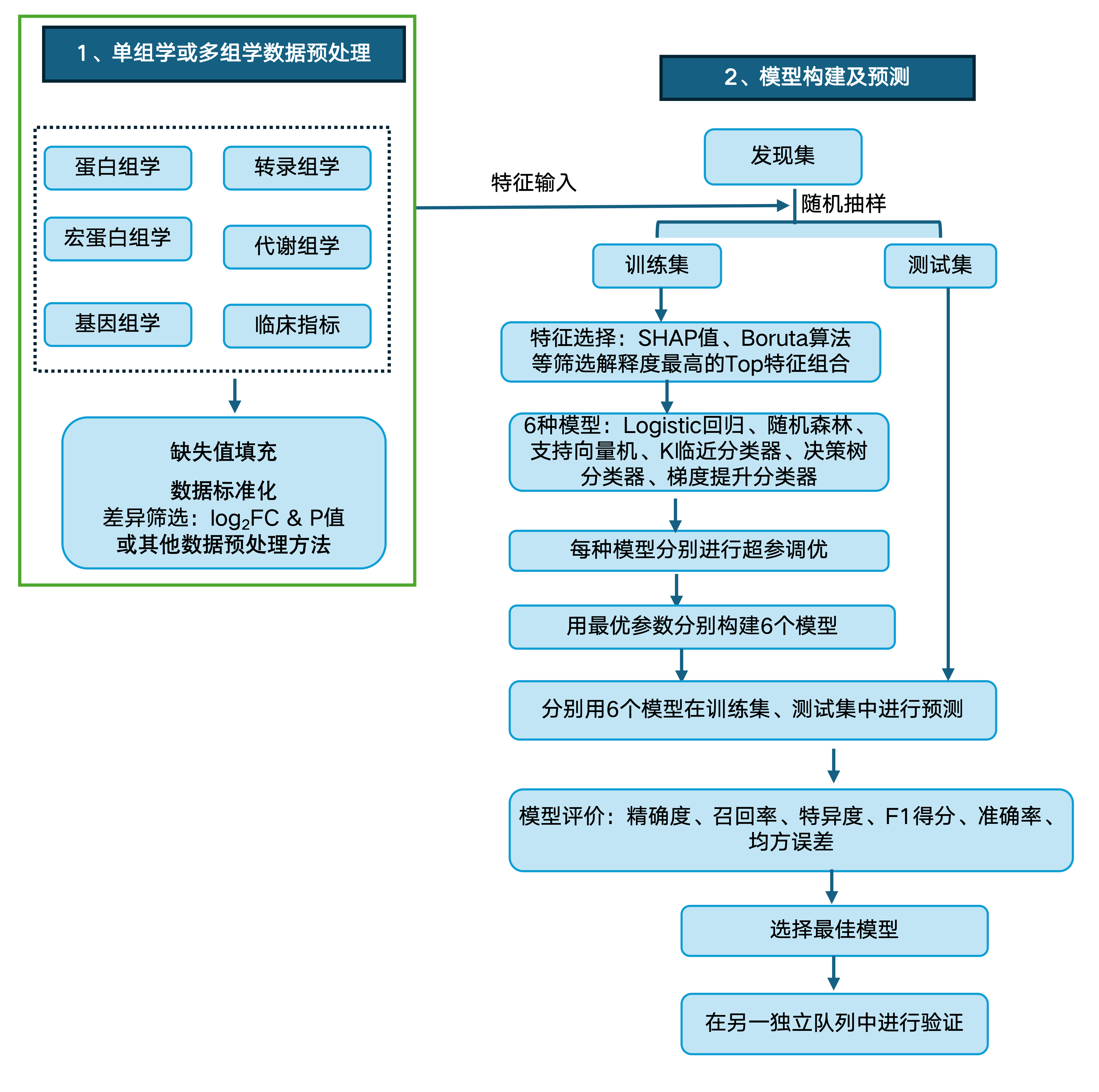
西湖欧米机器学习步骤包含严格的数据预处理、特征筛选、最优参数选择、多种机器学习集成、多种模型评价指标等。
严格的数据预处理确保数据质量,减少噪声和偏差的影响。特征筛选不仅能提高模型的性能和效率,还能增强模型的可解释性和泛化能力。网格搜索结合交叉验证不仅优化了模型参数,还防止了过拟合,同样提高了模型的泛化能力。通过集成多种模型(如logistic回归、随机森林、支持向量机、K临近分类器、决策树分类器和梯度提升分类器等),可实现多视角、多层次的分析,有助于快速获得最佳效果,降低过拟合。
多种评价指标(如精确度、召回率、特异性、F1得分、AUC-ROC等)的综合应用,使得模型性能评估更加全面和可靠,为生物标志物筛选和疾病预测提供了坚实的技术保障。
西湖欧米机器学习思路↓
西湖欧米自成立以来,一直致力于临床蛋白质组学大队列研究。其中,在以机器学习方法对各类临床疾病生物标志物的相关挖掘方面,发表了多篇高分文章,积累了丰富的项目经验。
PART 2
西湖欧米临床大队列——
机器学习案例详解
01
客户文章
Risk stratification of papillary thyroid cancers using multidimensional machine learning
多维度机器学习辅助甲状腺乳头状癌风险分层

▲ 发表杂志:International Journal of Surgery (IF=15.3)
▲ 发表年份:2023
▲ 研究背景:甲状腺乳头状癌(PTC)是最常见的内分泌恶性肿瘤之一,具有不同的风险等级。然而,全球范围内PTC的术前风险评估仍然极具挑战。
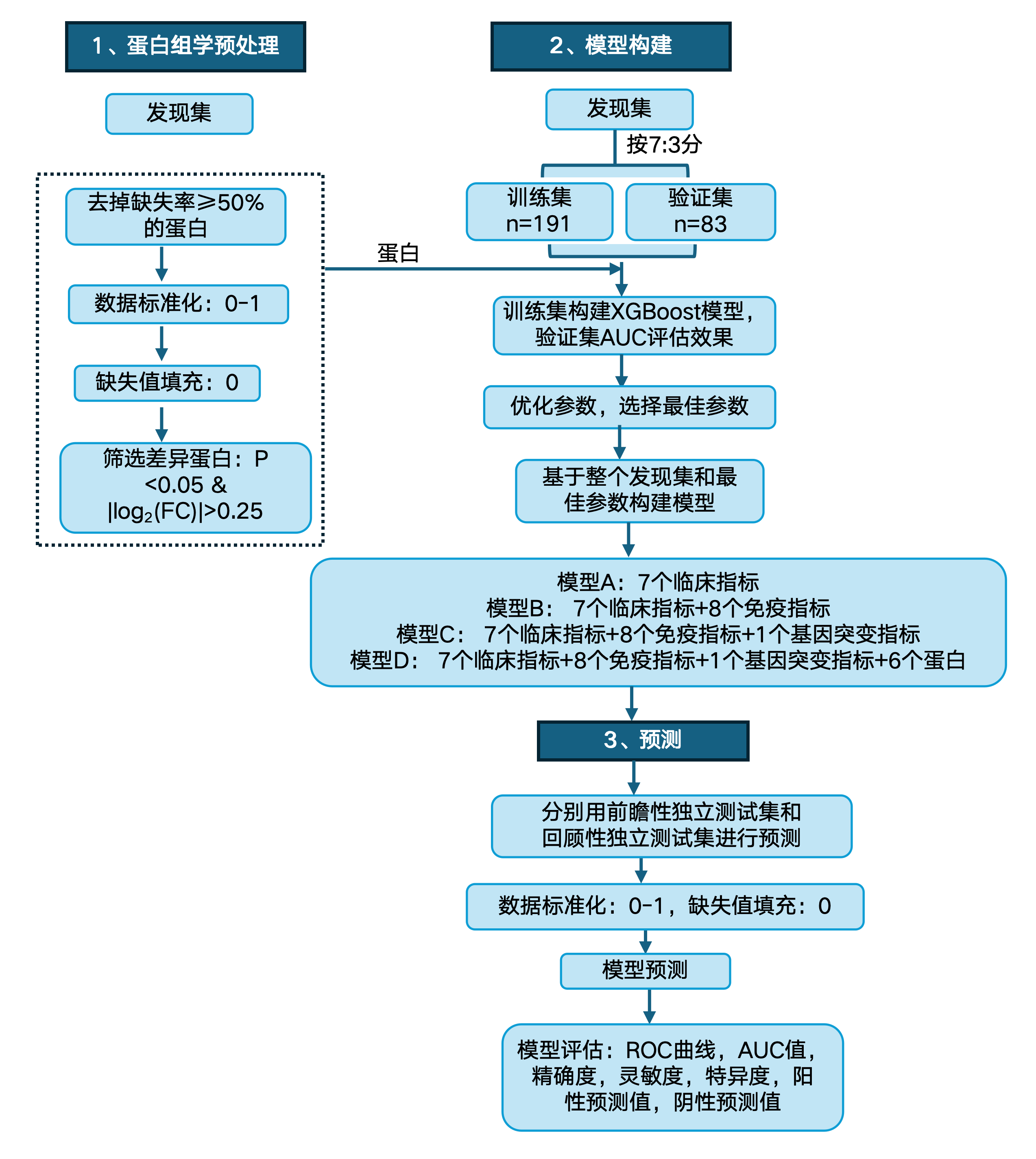
▲ 研究样本:研究者从多中心收集558例PTC临床样本,其中包含发现集(274例FFPE样本)、回顾性测试集(166例FFPE样本)和前瞻性测试集(118例FNA样本)
▲ 实验方法:数据非依赖的质谱采集技术(DIA-MS)
▲ 研究特色:机器学习(XGBoost)
▲ 研究结果:研究团队采用机器学习方法,利用蛋白质、基因突变、免疫和临床这四个维度信息成功构建甲状腺乳头状癌术前风险评估分类器(PRAC-PTC),能够良好区分低危和中高危甲状腺乳头状癌患者,并在回顾性和前瞻性研究中得到有效验证。
文章机器学习思路↓
文章机器学习关键结果:
基于XGBoost方法构建的模型进行预测,发现集(FFPE样本)中的AUC为0.925,回顾性独立测试集(FFPE样本)中的AUC为0.787,前瞻性独立测试集(细针穿刺样本)中的AUC为0.799。
基于蛋白质、基因、免疫和临床这四个维度信息构建的甲状腺乳头状癌术前风险评估分类器无论在回顾性测试集中还是前瞻性测试集中都优于模型A(基于临床指标)、模型B(基于临床和免疫指标)、模型C(基于临床、免疫和基因突变)。
模型在独立测试集中的性能评估↓

文章机器学习特色
★ 严格的数据预处理
★ 超参优化,选择最优参数
★ 多个独立测试集进行验证
★ 多项指标进行模型性能评估
★ 四种模型比较,选择最佳预测模型
02
客户文章
Integrated multiomic profiling of breast cancer in Chinese population reveals patient stratification and therapeutic vulnerabilities
整合中国人群乳腺癌多组学谱揭示患者分层和疗法易感性

▲ 发表杂志:Nature Cancer(IF=22.7)
▲ 发表年份:2024
▲ 研究背景:分子表达谱可以指导乳腺癌的精准治疗。目前大规模组学研究主要集中于基因组和转录组数据,然而,这些研究缺乏对下游生物过程的全面调查,并且未能利用更先进的方法,如多模态整合,以最大化其临床价值。虽然几项蛋白基因组学研究更新了我们对乳腺癌分子特征的理解,但其样本量通常限制在约100个,难以对特定亚型进行深入研究。此外,大多数数据基于西方人群,特别是白人个体,亚洲患者在公开数据集中代表性不足,亟待探索。
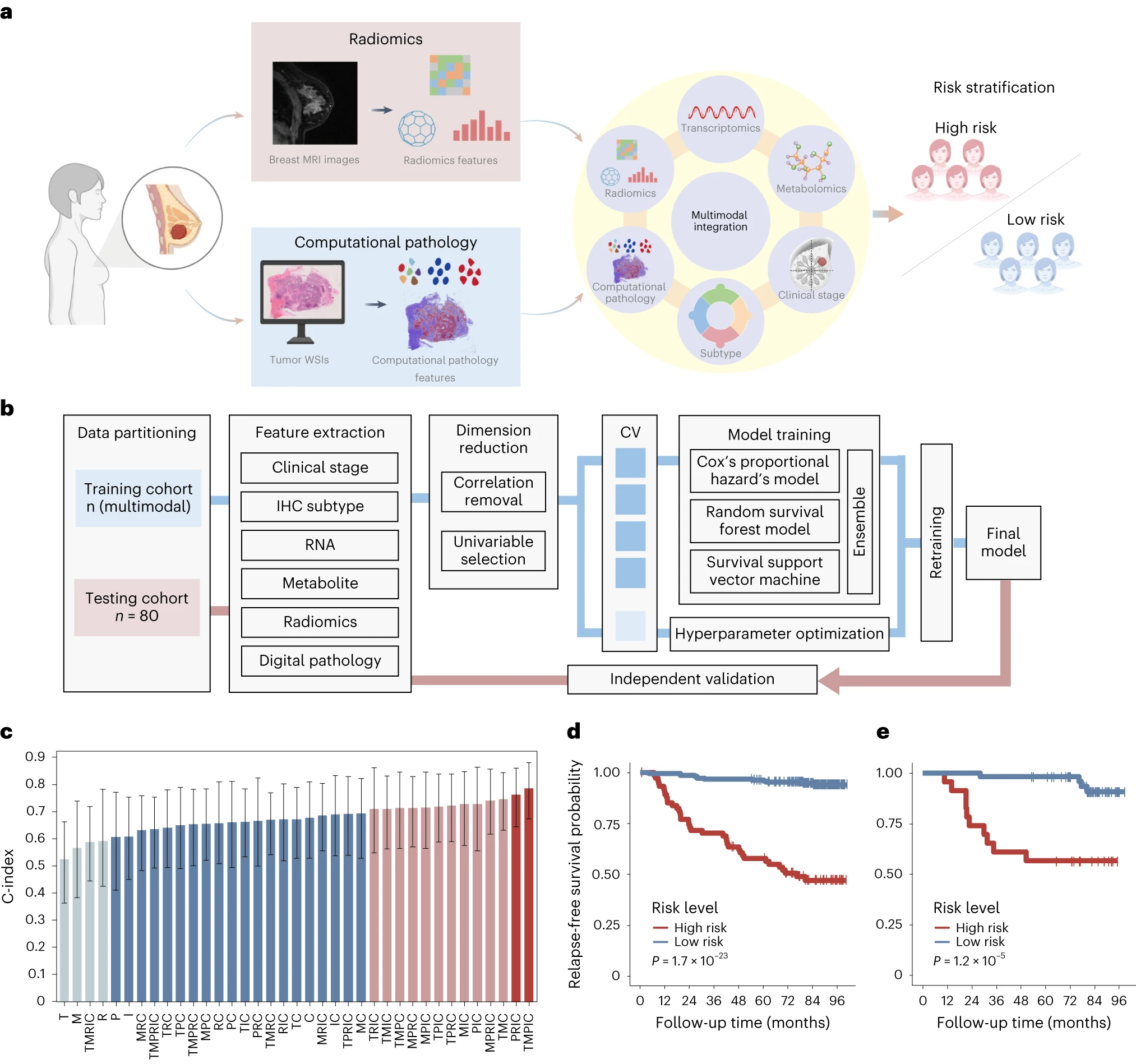
▲ 研究样本:研究纳入了2013-2014年期间在复旦大学附属肿瘤医院接受治疗的来自全国的773名乳腺癌患者(中位随访时间为 83.1个月),并分析了其基因组学、转录组学、蛋白质组学和代谢组学等多组学数据。
▲ 实验方法:同位素标记的串联质谱标记(TMT)
▲ 研究特色:分子分型+多组学整合分析+机器学习(Cox比例风险回归、随机森林、支持向量机)
▲ 研究结果:基于中国乳腺癌基因组图谱项目(CBCGA),文章研究了中国乳腺癌患者的临床和分子特征,包括肿瘤微环境、基因突变、多组学数据及其相互关系,并利用机器学习模型进行了预测和分类,为了解乳腺癌的病理特征、临床表现和治疗策略提供了重要的线索和数据支持。
文章机器学习关键结果↓
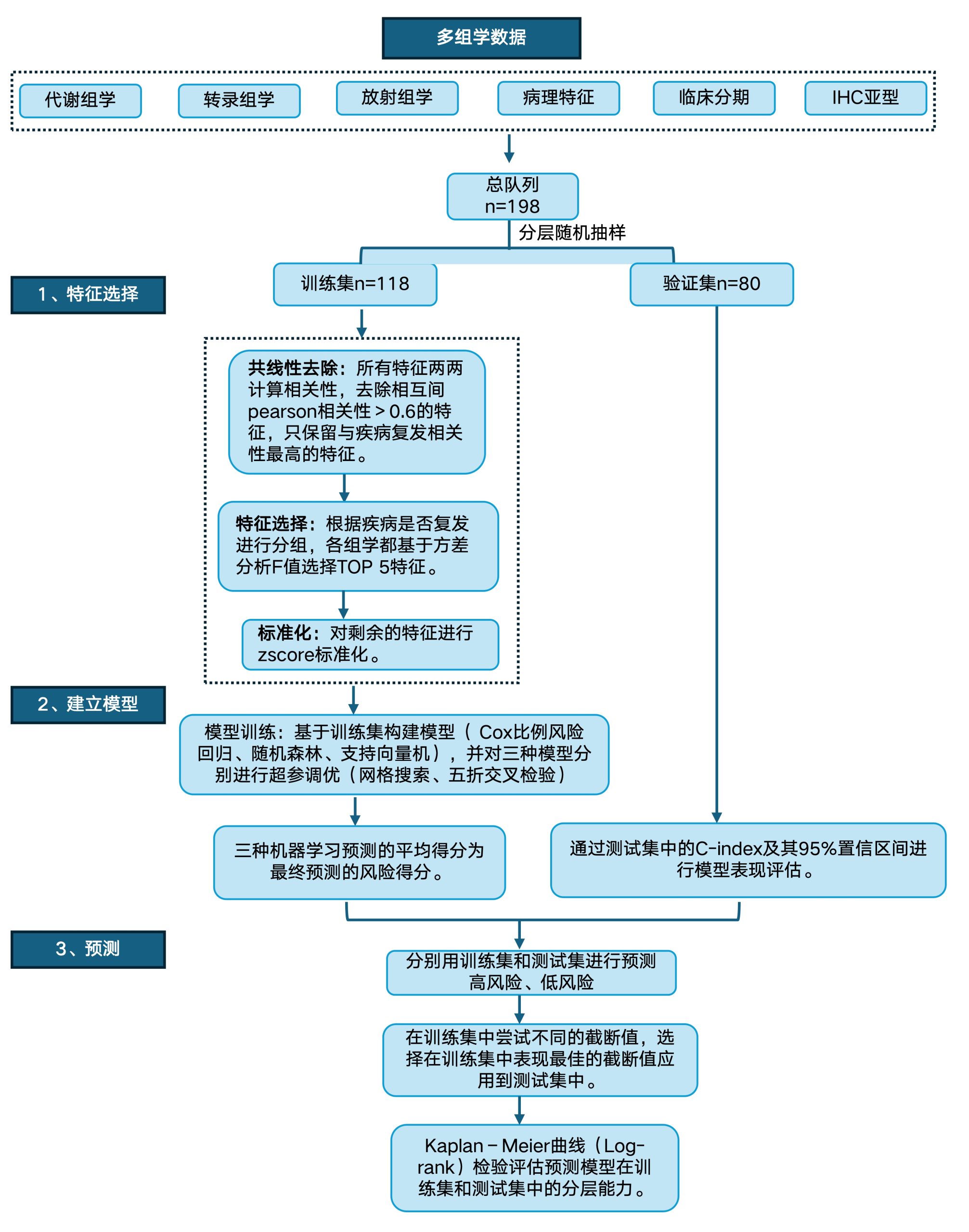
通过整合多模态(代谢组学、转录组学、放射组学、病理特征、临床分期、IHC亚型),基于Cox比例风险回归、随机森林、支持向量机三种模型的平均预测得分,可以很好地将患者分为高风险和低风险两组,且在训练集和测试集中两组的预后都有显著差异。
基于多组学的模型构建及风险等级预测↓
文章机器学习特色
★ 分层随机抽样,使样本更具代表性
★ 多组学数据整合,显著提高模型预测能力
★ 共线性去除,避免模型过拟合
★ 特征标准化,有效提升模型稳定性和准确性
★ 超参优化,选择最优参数
★ 三种机器学习方法构建模型,提高预测结果的准确性
★ 多种方法评估模型性能
03
合作文章
The Innovation| Alzheimer’s disease early diagnostic and staging biomarkers revealed by large-scale cerebrospinal fluid and serum proteomic profiling
通过大规模脑脊液和血清蛋白质组学分析揭示阿尔兹海默病早期诊断和分期生物标志物(2024,IF=32.1)

该研究纳入4个临床中心的221个研究对象的脑脊液样本和3个临床中心的288个研究对象的血清样本,通过随机森林模型构建了一个包含19个脑脊液蛋白和一个包含8个血清蛋白的生物标志物组合,AUC分别为0.984和0.881,能够准确地将阿尔兹海默病引起的轻度认知障碍者和正常认知者区分开来。
脑脊液及血清样本蛋白生物标志物的筛选↓

04
合作文章
Cell Reports Medicine| Population serum proteomics uncovers a prognostic protein classifier for metabolic syndrome
人群血清蛋白质组学揭示了代谢综合征的预后蛋白质分类器(2023,IF=14.3)

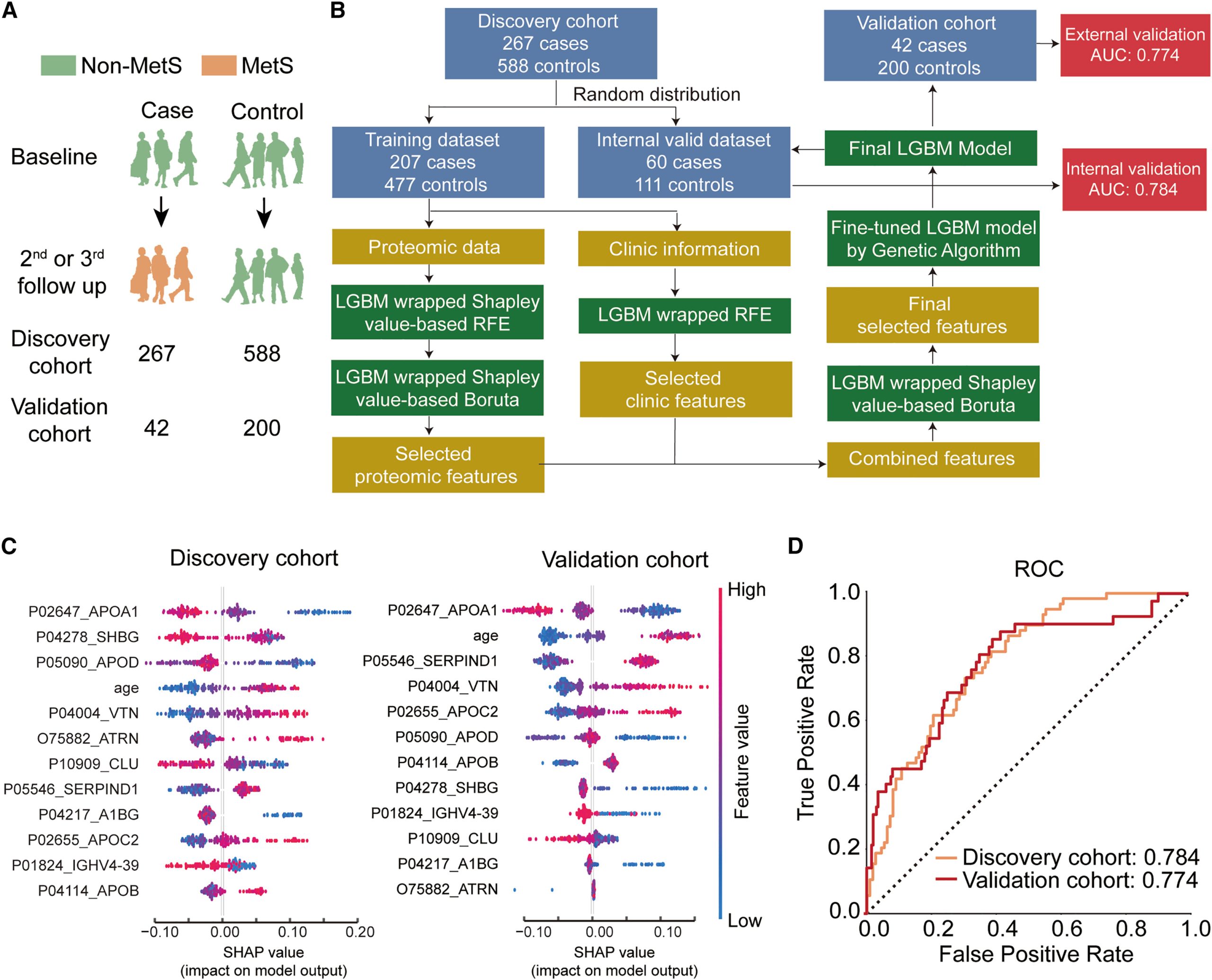
该研究发现队列共纳入855个研究对象,验证队列共纳入242个研究对象。研究人员通过LGBM模型构建了基于11个蛋白和年龄的生物标志物组合,在内部验证集和外部验证集中的AUC分别为0.774和0.784,揭示了血清蛋白在预测个体未来10年内发展为代谢综合征风险的能力。
基于蛋白质组学的代谢综合征预测模型构建及评估↓
05
合作文章
Microbiome| In-depth metaproteomics analysis of tongue coating for gastric cancer: a multicenter diagnostic research study
胃癌舌苔样本深度宏蛋白组学分析:一项多中心诊断研究(2024,IF=15.5)

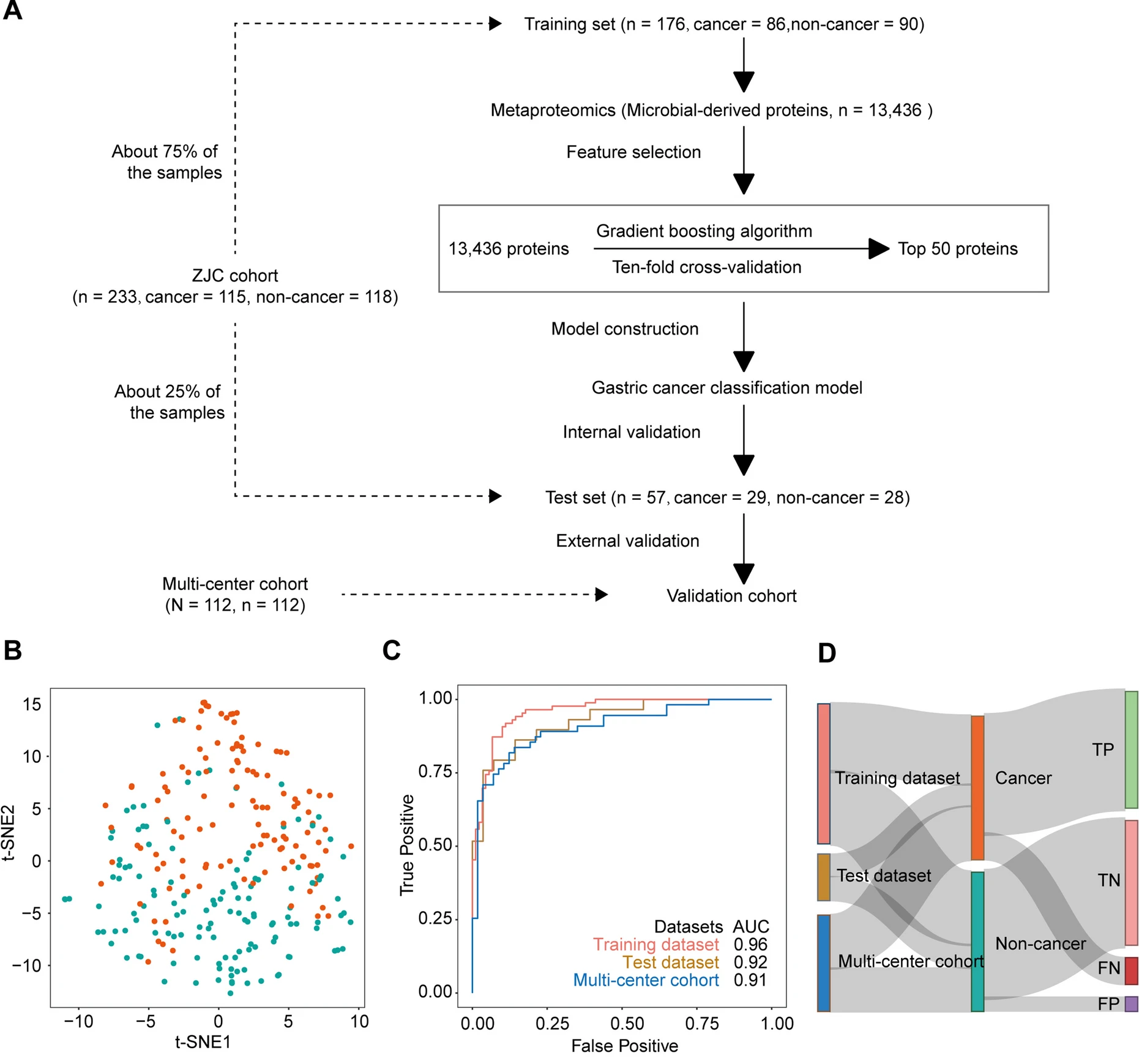
该研究发现队列共纳入233个研究对象,多中心验证队列共纳入112个研究对象。研究人员基于随机梯度提升算法构建了一个包含50个舌苔微生物蛋白的机器学习模型,在训练集、测试集、多中心验证集中的AUC分别达到了0.96、0.92、0.91,可以准确区分胃癌患者和非胃癌患者。
舌苔微生物蛋白的机器学习模型构建及评价↓
06
合作文章
Cell Discovery | Artificial intelligence defines protein-based classification of thyroid nodules
人工智能定义了基于蛋白质的甲状腺结节分类(2022,IF=33.5)

该研究发现队列共纳入578个研究对象,回顾性验证队列共纳入3个中心的271个研究对象,前瞻性验证队列共纳入9个中心的284个研究对象。研究人员开发了一个包含19个蛋白神经网络模型,在多中心的回顾性和前瞻性临床队列中AUC分别为0.94、0.93,可以准确区分良恶性甲状腺结节。
基于蛋白质组学的良恶性甲状腺结节预测模型构建及评价↓

PART 3
西湖欧米临床大队列研究——
机器学习常见QA
Q1:建议每组多少样本量?
A1:做机器学习,建议每组100例以上,越多越好。样本量越多,模型越准确。
Q2:什么是超参调优?
A2:超参调优(Hyperparameter Tuning)是指在机器学习和深度学习模型中,选择和调整模型的超参数(Hyperparameters)以优化模型性能的过程。常见的超参数包括学习率(Learning Rate)、正则化参数、批大小(Batch Size)、隐藏层的数量和神经元数量等。
它的策略就是:不断变换参数值,一轮一轮地去 “试”,直到找出结果最好的一组参数。其中网格搜索(Grid Search)和随机搜索(Randomized Search)是最基础也是最常用的两个。超参调优是机器学习和深度学习模型开发中的重要环节,合理的超参调优可以显著提升模型的性能和泛化能力。
Q3:什么是C-index,有什么作用?
A3:C-index,C指数,即一致性指数(concordance index),用来评价模型的预测能力。C指数是指所有病人对子中预测结果与实际结果一致的对子所占的比例。它估计了预测结果与实际观察到的结果相一致的概率。
Q4:什么是五折交叉检验,有什么作用?
A4:五折交叉检验(5-fold cross-validation)是一种评估机器学习模型性能的技术。它将数据集分成五个部分(称为“折”),然后进行多次训练和验证以得到模型的平均性能。
五折交叉检验的步骤
→ 划分数据集:将数据集随机分成五个大小相似的子集(折)。
→ 训练和验证:进行五次训练,每次使用其中四个折的数据来训练模型,剩下的一个折的数据用于验证模型。在每次训练和验证过程中,确保每个折都作为验证集使用一次。
→ 计算平均性能:将五次验证的性能指标(如准确率、精度、召回率、C指数等)进行平均,得到模型的总体性能评估。
五折交叉检验的作用:
→ 减少过拟合风险:通过在不同的训练和验证集上多次评估模型,五折交叉检验可以减少单次分割数据带来的偶然性,降低模型过拟合的风险。
→ 提供更可靠的性能评估:相比于单次训练/验证划分,五折交叉检验能够提供更稳定和可靠的模型性能估计。
→ 充分利用数据:五折交叉检验利用了所有数据进行训练和验证,最大化了数据的使用效率。
→ 模型选择和调参:在模型选择和超参数调优过程中,五折交叉检验可以作为评估标准,帮助选择性能更好的模型和超参数。
Q5:Boruta特征选择方法具体是怎么筛选特征的,有什么优势?
A5:Boruta特征选择方法是一种基于随机森林算法的特征选择方法,用于确定哪些特征对预测变量具有重要性。Boruta算法的核心思想是通过随机森林算法来评估特征的重要性,然后选择那些对预测结果有显著贡献的特征。它的独特之处在于引入了 “影子特征”(shadow features),这些影子特征是通过随机打乱原始特征生成的。
主要步骤
→ 生成影子特征:对于每个特征,生成一个影子特征。这些影子特征是通过打乱原始特征的顺序来创建的,因此它们与目标变量没有任何关系。
→ 构建随机森林模型:使用原始特征和影子特征一起训练一个随机森林模型。随机森林模型会计算每个特征(包括影子特征)的重要性得分。
→ 比较特征重要性:将原始特征的重要性得分与影子特征的重要性得分进行比较。具体来说,Boruta会将原始特征的重要性得分与所有影子特征中最高的重要性得分进行比较。
决策规则
根据比较结果,决定每个原始特征是否重要
→ 如果原始特征的重要性得分显著高于影子特征的最高重要性得分,则认为该特征是重要的。
→ 如果原始特征的重要性得分显著低于影子特征的最高重要性得分,则认为该特征是不重要的。
→ 如果原始特征的重要性得分介于两者之间,则暂时认为该特征的重要性不确定,继续进行更多的迭代。
迭代过程:Boruta会多次迭代上述过程,通过多次比较来确定每个特征的重要性,直到所有特征都被确定为重要、不重要或达到预定的最大迭代次数。
优势
→ 鲁棒性:由于Boruta使用了随机森林模型,并且引入了影子特征,它在处理高维数据和多重共线性问题上具有较高的鲁棒性。
→ 自动化:Boruta是一个自动化的特征选择方法,可以在不需要人工干预的情况下自动选择重要特征。
→ 全面性:它通过多次迭代和比较,能够更加全面地评估每个特征的重要性,避免了遗漏重要特征的风险。
应用场景:
→ Boruta特征选择方法广泛应用于各种机器学习任务中,尤其是在处理高维数据集(如基因组数据、文本数据)时,能够有效地减少特征数量,提高模型的性能和可解释性。
→ 通过使用Boruta特征选择方法,研究人员可以更加准确地识别出哪些特征在预测目标变量时具有显著的影响,从而构建更加精确和可靠的预测模型。
Q6:机器学习方法多种多样,如logistic,lasso,随机森林,XGBoost,如何进行选择?
A6:选择合适的机器学习模型是一个系统化的过程,需要考虑数据特性、任务需求、模型性能、解释性、计算资源和时间等多个因素,一般可以在训练过程中尝试多个模型,选择性能最好的。
确定问题类型
→ 分类问题:目标变量是离散的,如二分类问题(是否患病)或多分类问题(预测疾病的轻中重、早中晚期等)。
→ 回归问题:目标变量是连续的,如身高、体重。
数据特性分析
高维数据:Lasso回归、随机森林、XGBoost因为它们具有内置的特征选择能力。
低维数据:大多数模型都适用。
常见模型及其适用情况
→ 线性模型:线性回归、Logistic回归
优点:简单、易解释、训练速度快。
缺点:对非线性关系捕捉能力弱。
适用情况:特征与目标变量关系近似线性,数据量适中。
→ 正则化线性模型:Lasso回归、Ridge回归
优点:自动特征选择,防止过拟合。
适用情况:高维数据,特征数量多于样本数量。
→ 树模型:决策树、随机森林、梯度提升树(如XGBoost、LightGBM)
优点:捕捉复杂非线性关系,处理缺失值能力强,特征重要性解释。
缺点:复杂度高,训练时间长。
适用情况:大数据量,高维数据,特征间关系复杂。
→ 支持向量机(SVM)
优点:适用于高维空间,处理非线性关系。
缺点:对大规模数据训练速度慢。
适用情况:中小数据量,高维数据,分类问题。
→ 神经网络和深度学习模型:MLP、CNN、RNN等
优点:强大的非线性拟合能力,适用于图像、语音、文本等复杂数据。
缺点:需要大量数据和计算资源,难以解释。
适用情况:大数据量,复杂任务,如图像分类、自然语言处理。