






随着生物技术的快速发展,组学数据迅猛积累,进入了“大数据”的时代。在这个背景下,人工智能(AI)的崛起,特别是机器学习和深度学习方法,正引领着组学数据的革命性应用。在过去的十年中,机器学习在组学研究中的广泛应用已经为现代医学和生物医学领域带来了深刻的变革。
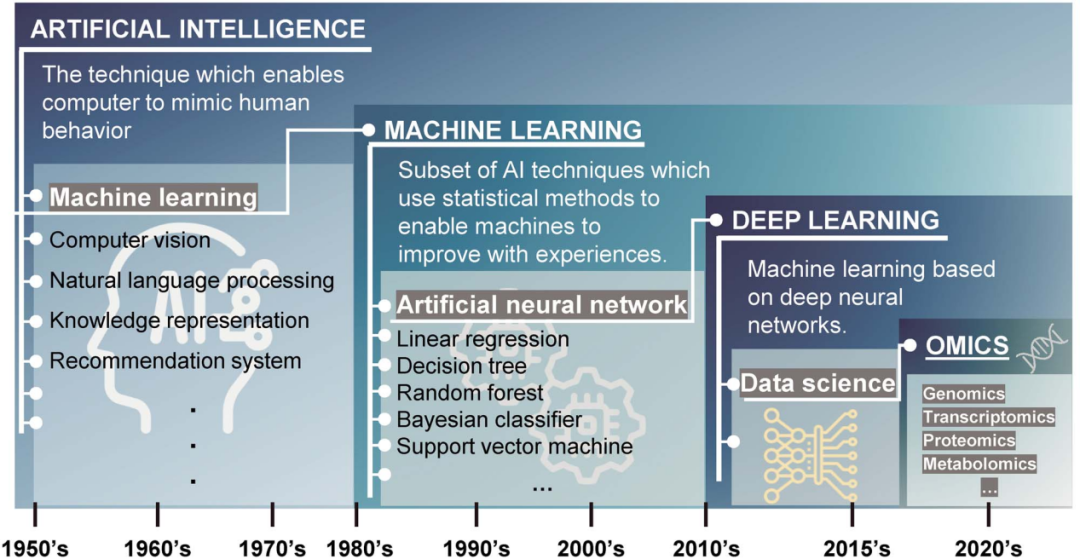
图1 人工智能、机器学习和深度学习 [1]
蛋白质组学是指研究基因组中表达的所有蛋白质及其特征,主要包括蛋白质结构、蛋白质丰度、蛋白质活性、蛋白质修饰、蛋白质定位、蛋白质相互作用等。
机器学习可被应用在处理蛋白质组质谱数据、筛选蛋白生物标志物、预测蛋白的基因结合位点及预测蛋白互作等多种蛋白质组学研究中。值得关注的是,机器学习在基于蛋白质组学的临床疾病标志物研究中的应用正日益引领着医学领域的未来。
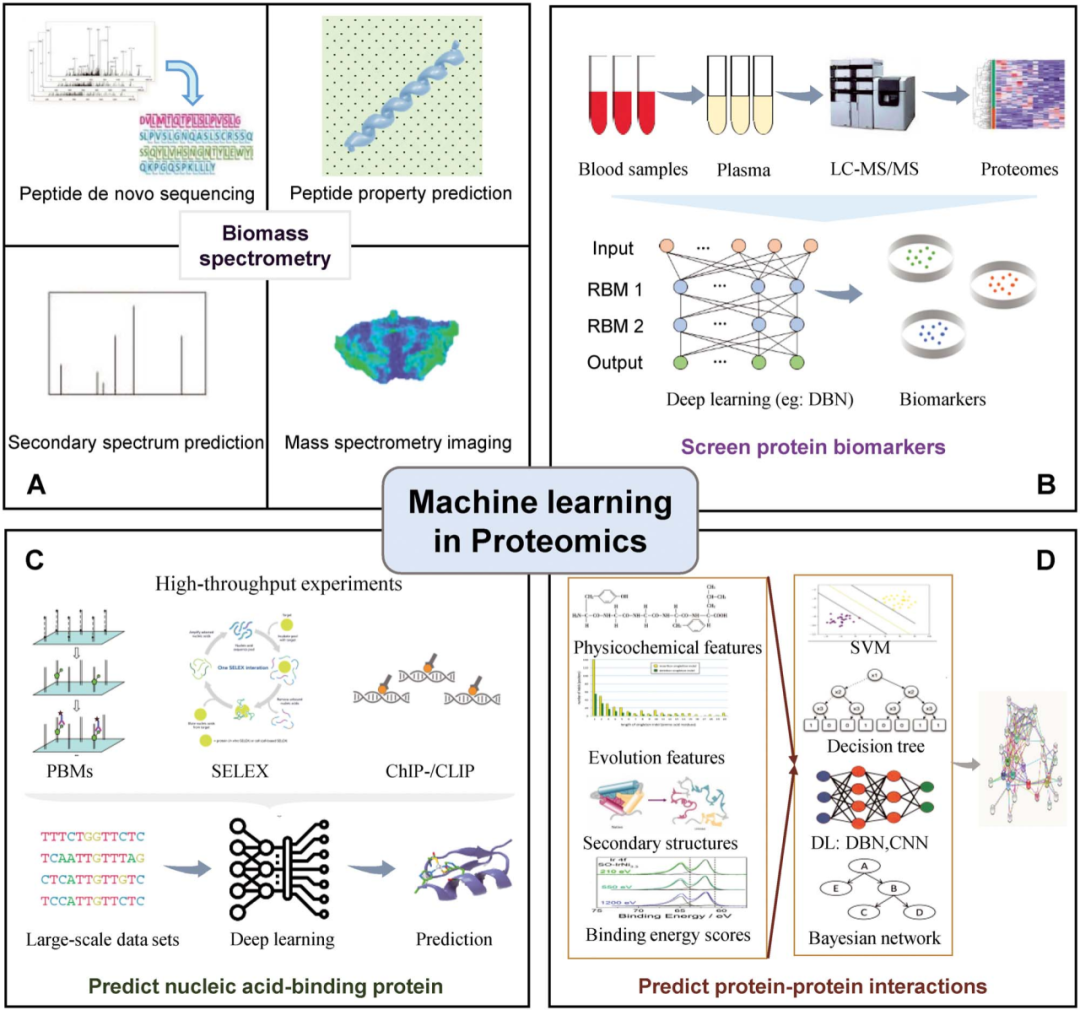
图2 机器学习在蛋白质组学中的应用[1]
在临床疾病标志物研究中,机器学习扮演着至关重要的角色,为以下方面的疾病生物标志物研究带来重大突破:
1. 疾病诊断:机器学习算法通过深度分析蛋白质组数据,能够准确识别与特定疾病或其亚型相关的生物标志物。这些标志物可以包括特定蛋白质的丰度、修饰、结构等多种特征。通过建立模型,机器学习帮助科学家确定哪些蛋白质或蛋白质组合在特定疾病的诊断和预测中具有高度相关性。
2. 疾病分类:机器学习方法利用蛋白质组数据,对不同疾病进行精确分类和诊断。通过训练算法使用已知疾病样本的蛋白质组数据,构建分类模型,使其能够识别未知样本的疾病状态。这对于制定个性化治疗策略至关重要。
3. 治疗反应预测:在临床治疗中,机器学习分析患者的蛋白质组数据,从而精确预测患者对不同治疗方案的反应。这有助于医生为患者选择最合适的治疗策略,提高治疗效果。
4. 疾病进展监测: 机器学习可定期分析患者的蛋白质组数据,实时监测疾病的进展和变化。这可用于评估治疗效果、调整治疗计划,以及早期发现疾病复发的迹象。
综合而言,机器学习在蛋白质组学数据挖掘中为临床疾病标志物的研究提供了强大的工具,为改善疾病的诊断、治疗策略提供强大助力。
西湖欧米专注于AI赋能的蛋白质组学,致力于以蛋白质组大数据技术创新为驱动力,利用机器学习挖掘临床标志物,目前已有多篇高分文章产出。
案例分享
案例1:
Cell Reports Medicine | 基于万例血清蛋白组预测代谢综合征风险[2]
文章概述:
2023年8月30日,西湖大学郭天南、郑钜圣及中山大学陈裕明教授,共同通讯在 Cell Reports Medicine 在线发表了题为 “Population serum proteomics uncovers a prognostic protein classifier for metabolic syndrome” 的研究论文。该研究基于一个随访10年以上的社区队列人群建立了近2万例血清蛋白质组学数据库,并构建了机器学习模型前瞻性预测代谢综合征在10年内的患病风险。

研究样本
(1)研究队列和样本:3840名参与者的7890个血清样本,并随机分为发现队列(n=4794)和验证队列(n=3094);
(2)组学方法:DIA蛋白质组;
对这些样本进行独立制备、质谱数据采集和数据分析。研究者在发现队列中检测到438种蛋白,在验证队列中检测到413种蛋白。利用这些数据,研究者构建了一个机器学习模型来评估血清蛋白质预测在十年内发生代谢综合征的风险的能力(图4B)。
最终模型的特征包括载脂蛋白A-I(APOA1)、性激素结合球蛋白(SHBG)、载脂蛋白D(APOD)、年龄、玻连蛋白(VTN)、吸引素(ATRN)、簇因子(CLU)、肝素辅因子2(SERPIND1,HCII)、α-1B-糖蛋白(A1BG)、载脂蛋白C-II(APOC2)、免疫球蛋白重链变量区域4-39(IGHV4-39)和载脂蛋白B-100(APOB)(图4C)。使用随机选择的内部验证数据集对模型进行测试,AUC为0.784,表明研究者的模型可以有效预测在十年内发生代谢综合征的风险(图4D)。
最后,研究者使用来自独立验证队列的242个样本进行模型测试,得到AUC为0.774(图4D)。这些结果表明所选特征是稳定的,可以很好地推广到独立采集的样本,并且在模型中使用的蛋白质可能作为有前景的生物标志物候选者。
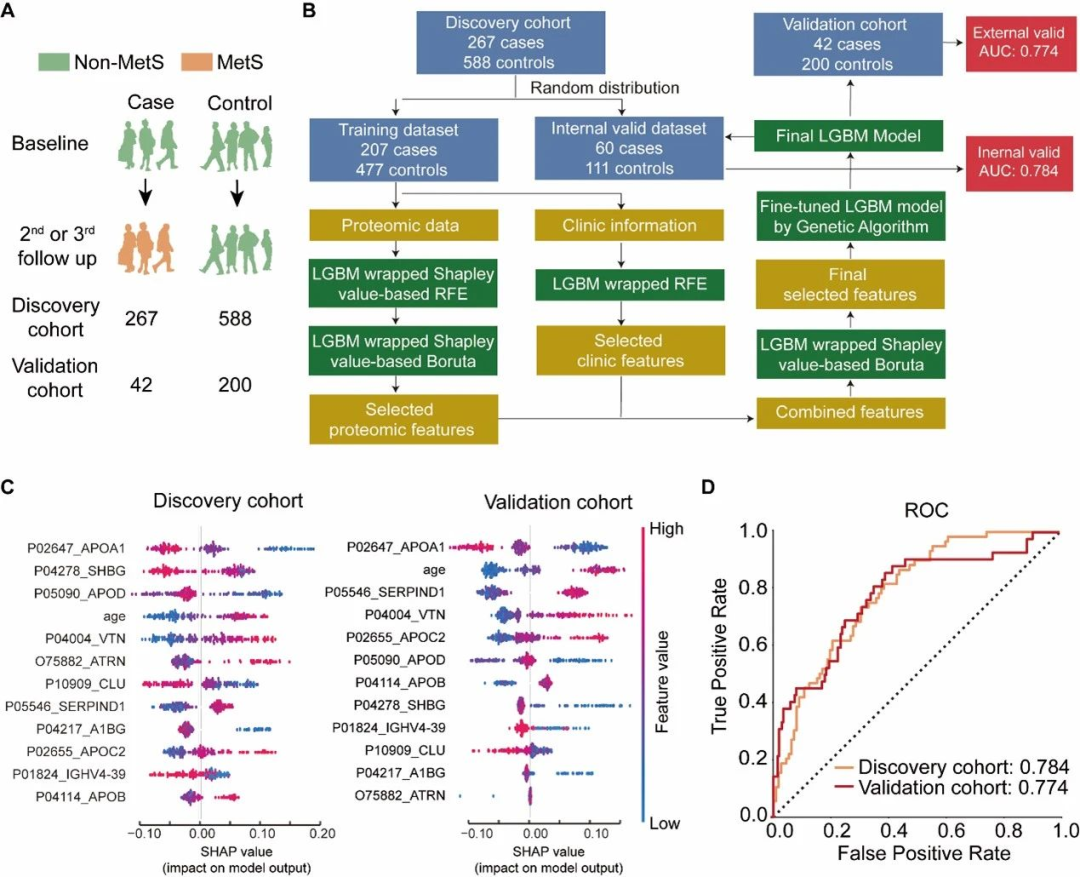
图4 机器学习模型构建流程和结果
案例2:
Cell Discovery | AI人工智能结合蛋白质组学辅助甲状腺结节良恶性判别[3]
文章概述:
2022年9月6日,西湖大学郭天南团队、李子青团队联合多个国内外临床单位共同在 Cell Discovery 发表了题为 “Artificial intelligence defines protein-based classification of thyroid nodules” 的文章。该研究通过人工智能(Artificial intelligence,AI)算法结合蛋白质组特征构建了一个可以用于甲状腺结节良恶性评估的深度学习模型。

研究样本
(1)研究队列和样本:
发现集:578名患者的579例甲状腺结节样本;
测试集:回顾性测试集(271例患者的288例FFPE样本)和前瞻性测试集(294例FNA活检样本);
(2)组学方法:DIA蛋白质组;
为建立一个基于蛋白质特征的用于区分甲状腺结节良恶性判断的AI模型,研究者首先从discovery set数据集的579例甲状腺样本中,通过遗传算法结合神经网络模型进行蛋白特征选择,之后对自主构建模型进行训练,并在独立测试集中对模型验证。流程如下:先将发现数据集(n = 579)随机分为数据集A,包含2/3的样本(n = 386),其余样本构成数据集B(n = 193),接着通过遗传算法结合3折交叉验证从数据集A中选择蛋白特征组合。通过遗传算法得到19个蛋白组合,进一步在数据集B中验证对甲状腺良恶性结节判别的准确率,并选取准确率最高的一组作为最终的19个蛋白组合,最后通过SHapley Additive exPlanations(SHAP)算法评估19个蛋白质特征对分类器的重要性并排序。通过该方法筛选到的19个关键蛋白全部直接或间接的与甲状腺疾病或功能相关,表明该方法在挖掘与甲状腺结节良恶性区分有关的潜在蛋白方面非常具备潜力。
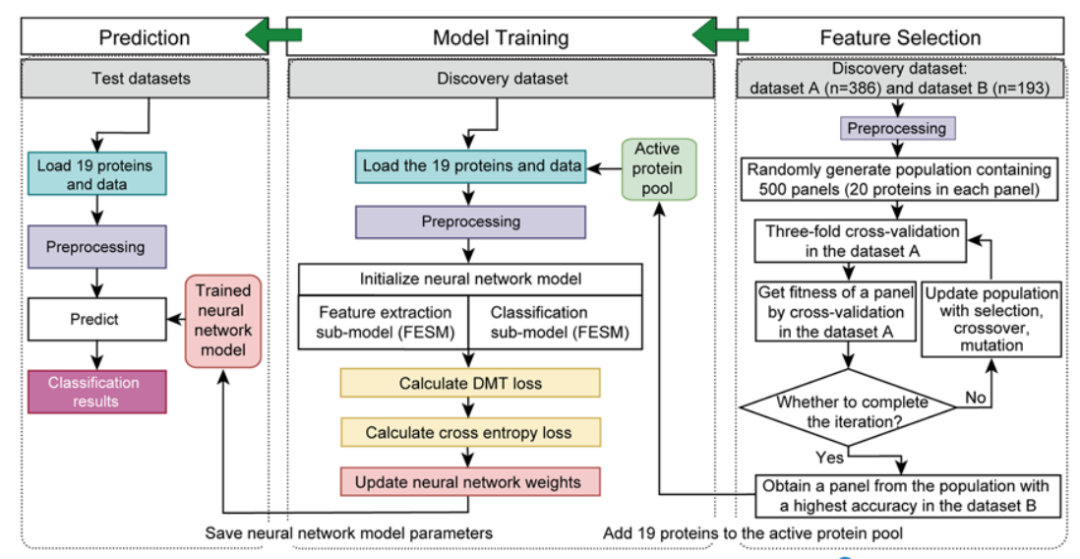
图6 机器学习模型构建流程
研究者进一步对构建的模型进行效果评价,基于筛选出的19种蛋白质特征,研究者将自主构建的模型与其他6种不同的机器学习模型进行比较,结果表明研究者自主构建的模型具有最高的分类效能,AUC高达0.93。随即,研究者又在两个独立队列(回顾性队列和前瞻性队列)进行了验证,AUC分别为0.94和0.93,准确性分别为89%和85%。此外,从下UMAP图中可知,这19个蛋白对良恶性甲状腺结节可以进行明显区分。
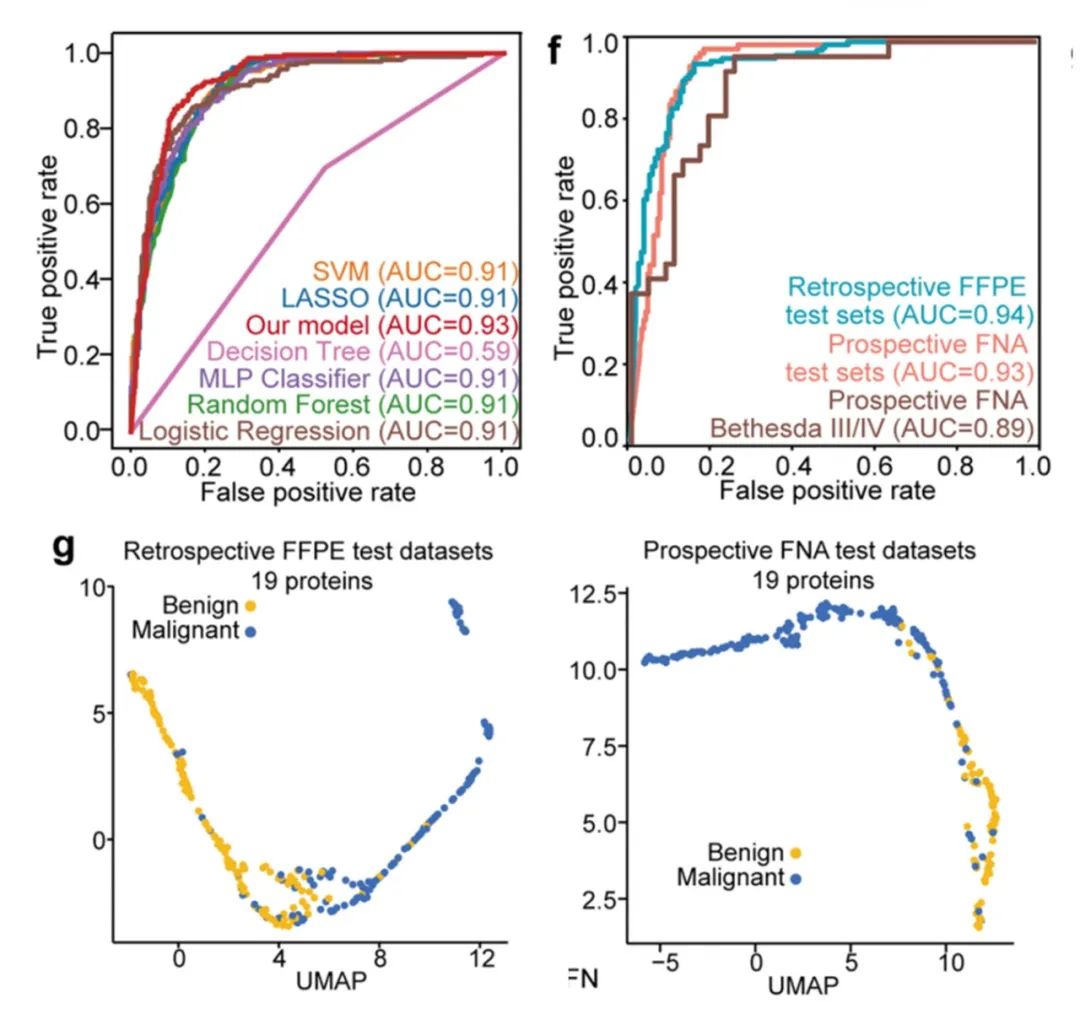
图7 模型性能测试和验证
西湖欧米临床疾病标志物研究套餐
在临床蛋白质组学研究上,西湖欧米积累了多年的高通量、高稳定性的蛋白质组学样本制备和数据分析经验,联合AI 深度学习形成了 AI 赋能的蛋白质组学技术平台。
西湖欧米为临床研究客户提供机器学习为核心的临床疾病标志物研究方案,为临床研究客户提高大队列样本的专业队列设计,支持极微量、多种临床组织样本的深度高通量蛋白质组学分析。
感兴趣的老师赶紧联系我们吧。

参考文献:
[1] Li R, Li L, Xu Y, et al. Machine learning meets omics: applications and perspectives. Brief Bioinform. 2022 Jan 17;23(1):bbab460.
[2] Cai X, Xue Z, Zeng FF, et al. Population serum proteomics uncovers a prognostic protein classifier for metabolic syndrome. Cell Rep Med. 2023 Sep 19;4(9):101172.
[3] Sun Y, Selvarajan S, Zang Z, et al. Artificial intelligence defines protein-based classification of thyroid nodules. Cell Discov. 2022 Sep 6;8(1):85.


