






Introduction
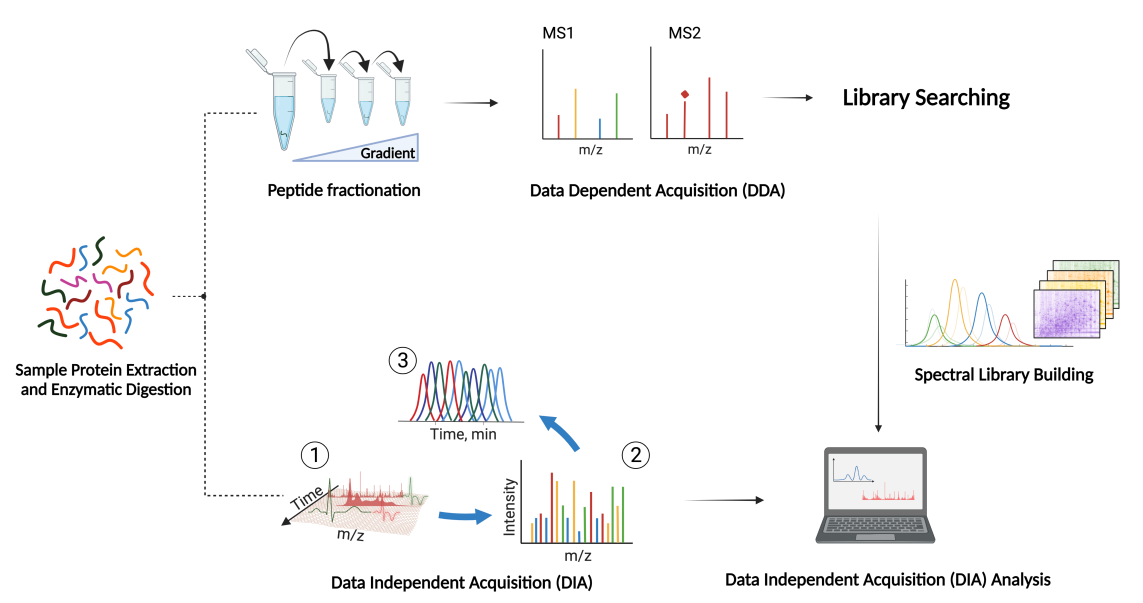
DIA (data-independent acquisition) is a non-discriminatory and non-random proteome analysis technology. It divides the full scanning range of mass spectrometry into several windows, and then detects and breaks all ions in each window. This is to obtain information on all ions in the sample, reduce the missing value of sample detection, and improve the quantitative accuracy and repeatability, achieving highly stable and accurate proteome quantitative analysis in a large sample queue.
Services
- 1. By combined with PCT sample pre-processing technology, Westlakeomicscan achieve high depth protein quantitative analysis of clinical micro samples (such as FFPE, puncture biopsy, tears, etc.), and the minimum sample delivery volume of tissue samples is only 0.1mg.
- 2. Use a variety of proteomics database search software, including OpenSWATH, EncyclopeDIA, DIA-NN, etc., and be able to comprehensively analyze the results to improve the identification amount and quantitative accuracy of proteins.
Mass Spectrometer
Orbitrap Exploris 480
Project Case
In this study, we profiled the proteomic tissue landscape of CRC evolving from normal colon to hyperplastic polyps, adenomas, adenocarcinoma not otherwise specified (AC) or mucinous adenocarcinoma (MC). We identified 69,949 peptides, 6,359 protein groups, and 4,830 unique proteins based on our previously established spectral library for DIA analysis from 170 FFPE tissue samples (85 patients, each with 2 biological replicates). Pearson’s correlation coefficient between biological replicates was 0.813, and 0.953 between technical replicates.
References
2. Röst et al. OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nature Methods. 2016.13(9):741-748
https://www.nature.com/articles/nmeth.3959
3.Röst, et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nature Biotechnology. 2014.32:219-223
https://www.nature.com/articles/nbt.2841
4.Guo, et al. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nature Medicine. 2015.21(4):407–413.
https://www.nature.com/articles/nm.3807
5. Searle et al. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nature Communications. 2018. 9(1): 1-12
https://www.nature.com/articles/s41467-018-07454-w
6.Xu, et al. In-depth Serum Proteomics Reveals Biomarkers of Psoriasis Severity and Response to Traditional Chinese Medicine. Theranostics. 2019.9(9): 2475-2488.
https://www.thno.org/v09p2475.htm
7.Shao, et al. Comparative analysis of mRNA and protein degradation in prostate tissues indicates high stability of proteins. Nature Communications. 2019. 10(1):2524.
https://www.nature.com/articles/s41467-019-10513-5
8.Zhu, et al. High-throughput Proteomic analysis of FFPE tissue samples facilitates tumor stratification. Molecular Oncology. 2019 Sep;13(11): 2305-2328.
https://febs.onlinelibrary.wiley.com/doi/10.1002/1878-0261.12570
9.Zhang, et al. Data-Independent Acquisition Mass Spectrometry-Based Proteomics and Software Tools: A Glimpse in 2020. Proteomics. 2020.20(17-18): e1900276.
https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/pmic.201900276
10.Demichev, et al. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nature Methods. 2020.17:41-44
https://www.nature.com/articles/s41592-019-0638-x
11.Cai, et al. PulseDIA: Data-Independent Acquisition Mass Spectrometry Using Multi-Injection Pulsed Gas-Phase Fractionation. Journal of Proteome Research. 2021.20(1):279-288.
https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00381
12.Ge, et al. Computational Optimization of Spectral Library Size Improves DIA-MS Proteome Coverage and Applications to 15 Tumors. Journal of Proteome Research. 2021
https://pubs.acs.org/doi/full/10.1021/acs.jproteome.1c00640
13.Liu, et al. DIA-based Proteomics Identifies IDH2 as a Targetable Regulator of Acquired Drug Resistance in Chronic Myeloid Leukemia. Mol Cell Prot. available at bioRxiv, 2021.
https://www.mcponline.org/article/S1535-9476(21)00159-6/fulltext#secsectitle0030
14.Shao, et al. Proteomics profiling of colorectal cancer progression identifies PLOD2 as a potential therapeutic target. Cancer Commun. 2021.
https://onlinelibrary.wiley.com/doi/10.1002/cac2.12240
15.Zhu, et al. Snapshot: Clinical proteomics. Cell. 2021.184(18): 4840-4840.


