






胰腺导管腺癌(PDAC)是最具侵袭性的恶性肿瘤之一,传统的临床和分子标志物分析未能准确预测PDAC患者的临床结果。将肿瘤和宿主的全面分子评估与机器学习(ML)模型相结合,或可改善这一现状。
2024年1月22日,美国 Samuel Oschin 综合肿瘤研究所的 Dan Theodorescu、Jennifer E. Van Eyk 团队在 Nature Cancer 上发表了新的研究 The Molecular Twin artificial-intelligence platform integrates multi-omic data to predict outcomes for pancreatic adenocarcinoma patients。

图1 论文截图
文章介绍了一种新型的精准医学平台 Molecular Twin(“分子双胞胎”),该平台融合了宿主和肿瘤的多个分子、组织病理学和临床特征,以及基于机器学习的综合多组学分析(尤其是血浆蛋白组分析),可准确预测PDAC患者的疾病生存情况。
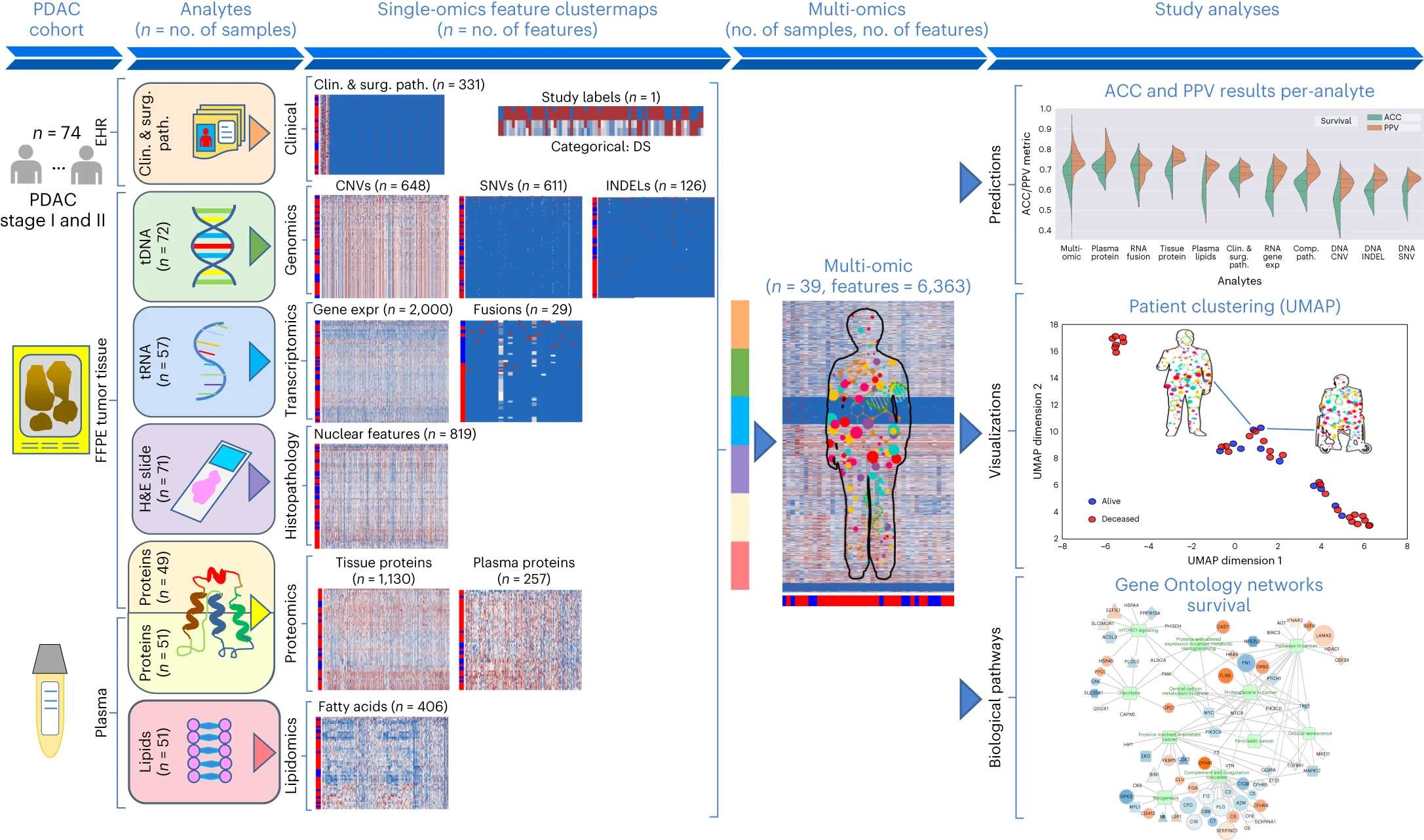
图2 Molecular Twin平台 对74名接受手术治疗的I/II期可切除PDAC患者的血浆和组织样本进行了高通量测序(NGS)、全转录组 RNA 测序、组织蛋白组学、血浆蛋白组学、脂质组学和计算病理学分析。这6,363个特征被合并,并作为七种不同类型的机器学习模型的输入,生成多组学生物标志模型,用于预测临床结果、提供患者级别的聚类数据,并洞察可能的治疗靶点。(EHR:电子健康档案)
结论1:
临床和手术病理特征影响结果
研究人员使用 “Molecular Twin Pilot(MT-Pilot)” 队列中74名手术切除的胰腺导管腺癌(PDAC)患者的数据,其中临床分期为I(n=47)和II(n=27),于2015年3月至2019年4月之间接受手术切除,研究采集了手术时的肿瘤标本和术前的血浆标本。
患者的基线特征和临床特征被纳入多组学分析,包括高通量测序(NGS)、全转录组 RNA 测序、组织蛋白组学、血浆蛋白组学、脂质组学和计算病理学。通过这些分析得到的特征用于验证单组学和多组学模型,用于预测患者的死亡状态。研究使用了交叉验证方法,并利用四个独立数据集进行验证。
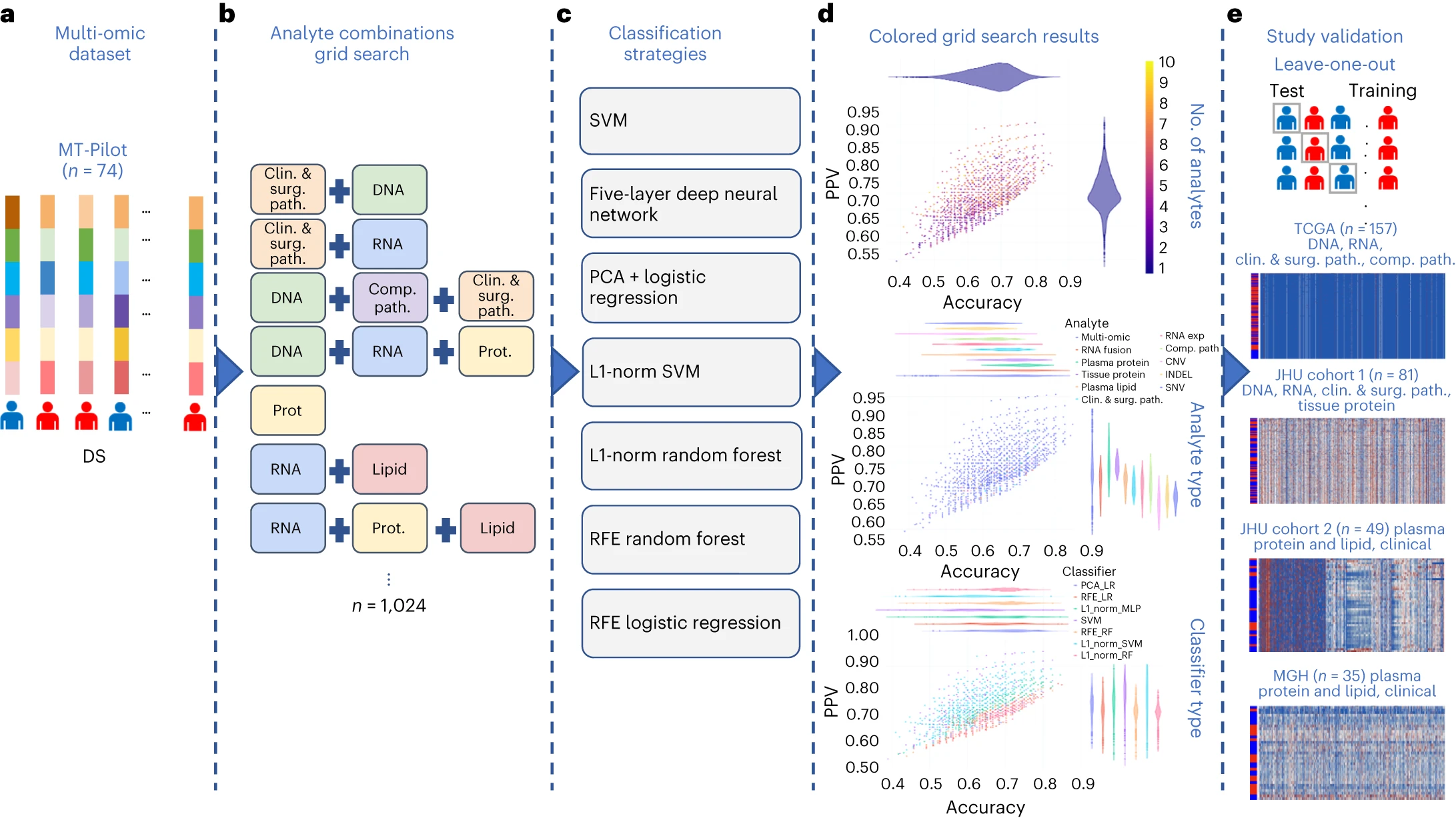
图3 研究分类方法概述 a:由 6,363 个经过处理的特征组成的多组数据集,涵盖临床和手术病理学、SNV、CNV、INDEL、RNA、fusion、组织蛋白、血浆蛋白、脂类和计算病理学分析物
研究人员应用多个机器学习模型分析了331个临床特征,包括手术病理特征和化疗治疗。其中,随机森林(RF)模型在确定疾病存活率(disease survival,DS)方面表现最佳,准确度为0.70,阳性预测值(PPV)为0.71。模型中关键特征包括合并症(如高脂血症、黄疸和胰腺炎),以及手术切缘状态等。DS模型主要受合并症的影响,占据了331个总特征中的306个。
结论2:
DNA 分析揭示了对预后具有重要意义的变化
通过DNA分析,研究人员揭示了与预后重要性相关的变化。在已知的PDAC致癌基因和抑癌基因中,点突变和插入/缺失(INDEL)多态性很常见。对611个体细胞单核苷酸变异(SNV)、648个拷贝数变异(CNV)和126个INDEL进行了组织样本处理。这些特征随后用于患者DS预测模型(附表3)。
RF模型在SNV分析中表现最佳,关键预测基因包括RAD51、IL6R、FGF20和SOX2等。CNV分析中,FOXQ1和KDM5D是DS相关的顶级预测因子。INDEL分析中,TP53、CDKN2A和SMAD4等基因突变与DS相关。
结论3:
药物抵抗的RNA特征影响预后
研究人员对74个福尔马林固定石蜡包埋(FFPE)肿瘤组织样本中的72个进行了全转录组测序。通过差异表达分析选择RNA基因转录本,采用L1归一化的RF模型,其预测DS的准确度为0.68。DS预测模型中NFE2L2和LRIG3基因是两个关键特征。在RNA融合的分析中,SVM模型预测DS的准确度为0.75。
结论4:
血浆蛋白是预测存活的关键生物标志物
研究人员利用蛋白质组学和脂质组学分析,最初生成了3777个肿瘤组织蛋白质、1051个血浆蛋白质和939个脂质组学特征。
对于组织蛋白质特征,预测DS的最佳模型是RF模型,准确度为0.73。而对于血浆蛋白质特征,最佳模型是五层深度神经网络模型(five-hidden-layer-deep neural network model),准确度为0.75。
其中,预测DS的血浆蛋白中包括ANXA1,该蛋白在胰腺癌发生和PDAC药物抵抗中起着重要作用。血浆脂质特征的RF模型预测DS的准确度为0.71,其中主要受到二酰基甘油和胆固醇酯的驱动。
结论5:
通过计算病理学预测细胞核形态
研究人员基于AI的计算病理学流程对71个H&E染色的PDAC 组织全切片图像(WSI)进行了评估。该流程包括两个卷积神经网络模型:一个用于遮盖癌细胞(mask-out cancer cells),另一个用于划分细胞核(delineate nuclei)。
通过特征定量分析核形态和纹理,发现一些特征具有预后潜力。通过留一法(leave-one-out)交叉验证,采用随机森林(RF)模型预测DS,准确度为0.66,PPV为0.76。同时,评估计算病理学预测DS是否受到样本中癌症百分比或癌症与基质比率的影响,结果未发现显著差异,但发现新辅助治疗后组织中基质的百分比显著增加。
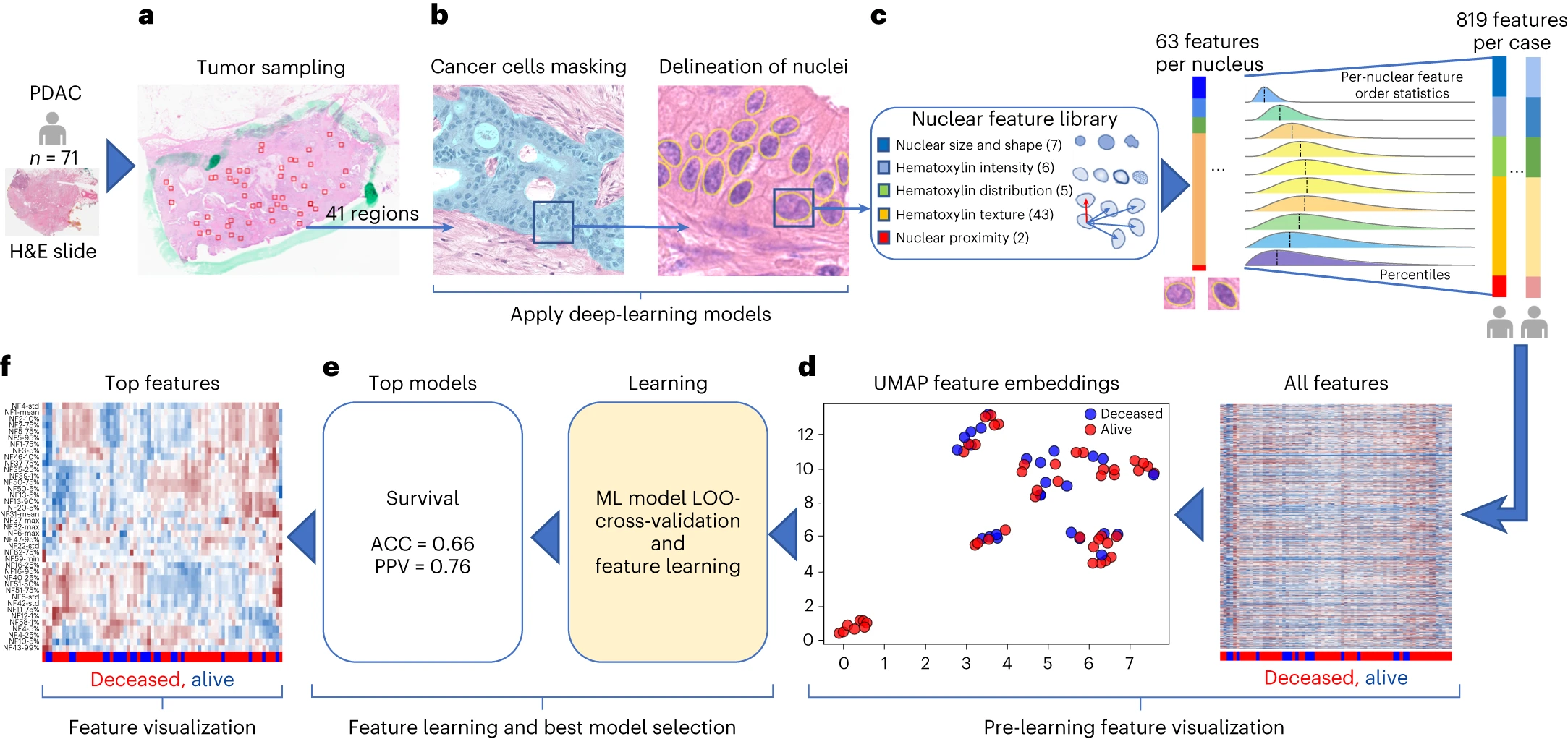
图4 计算病理学流程
结论6:
多组学分析优于单组学分析,揭示分层互补性
通过多组学分析,结合七个独立机器学习模型,研究人员对来自单一组学来源的6,363个特征进行了综合分析。最终得到的多组学模型在预测DS方面表现最佳,准确性达到0.85,PPV为0.87。
单一组学模型显示每个分析方法都具有一定的预测力,但多组学模型在准确性和PPV方面表现更佳,这表明在多组学分析中的组合方式呈现出互补性和信息增益。
通过排除特征的方法,研究人员对每个分析类别的重要性进行了量化,结果显示排除任何一个分析类别并不会显著改变模型的性能。前15个多组学模型中,血浆蛋白分析在特征贡献中占主导地位。
结论7:
多组学模型为深入了解PDAC提供了生物学依据
研究的探索目标之一是评估 Molecular Twin 平台是否能够识别PDAC潜在的治疗途径和靶点。
通过使用差异表达的特征集,研究人员对所有分子组学特征的重要性进行了评估,并生成了蛋白质、DNA 和 RNA 的本体可视化图表。在 PDAC 中,mTOR 信号通路是已知的通路之一,而且在多种肿瘤中都有作用。本研究的 Gene Ontology 网络可视化显示了 mTOR 信号通路。除了 mTOR 之外,Gene Ontology 网络可视化还揭示了 PDAC 中其他临床和生物学相关的通路,包括糖酵解和细胞代谢。
接着,研究人员通过使用所有分子标志物的6363个特征,根据多组学分子标志物的病人级别聚类,对病人的异质性与临床结果之间的关系进行了研究。聚类结果显示了三个簇,其中 Cluster 1 表示临床结果相同的患者(全部已故)。通过比较不同簇中某个特征的表达,发现了在每个分子组学分析中存在相关特征的比例。这些结果表明,该研究方法对于特定簇患者(例如 Cluster 1)的预测将变得越来越准确,同时对于哪些特征对于个体患者簇至关重要提供了更深入的了解。
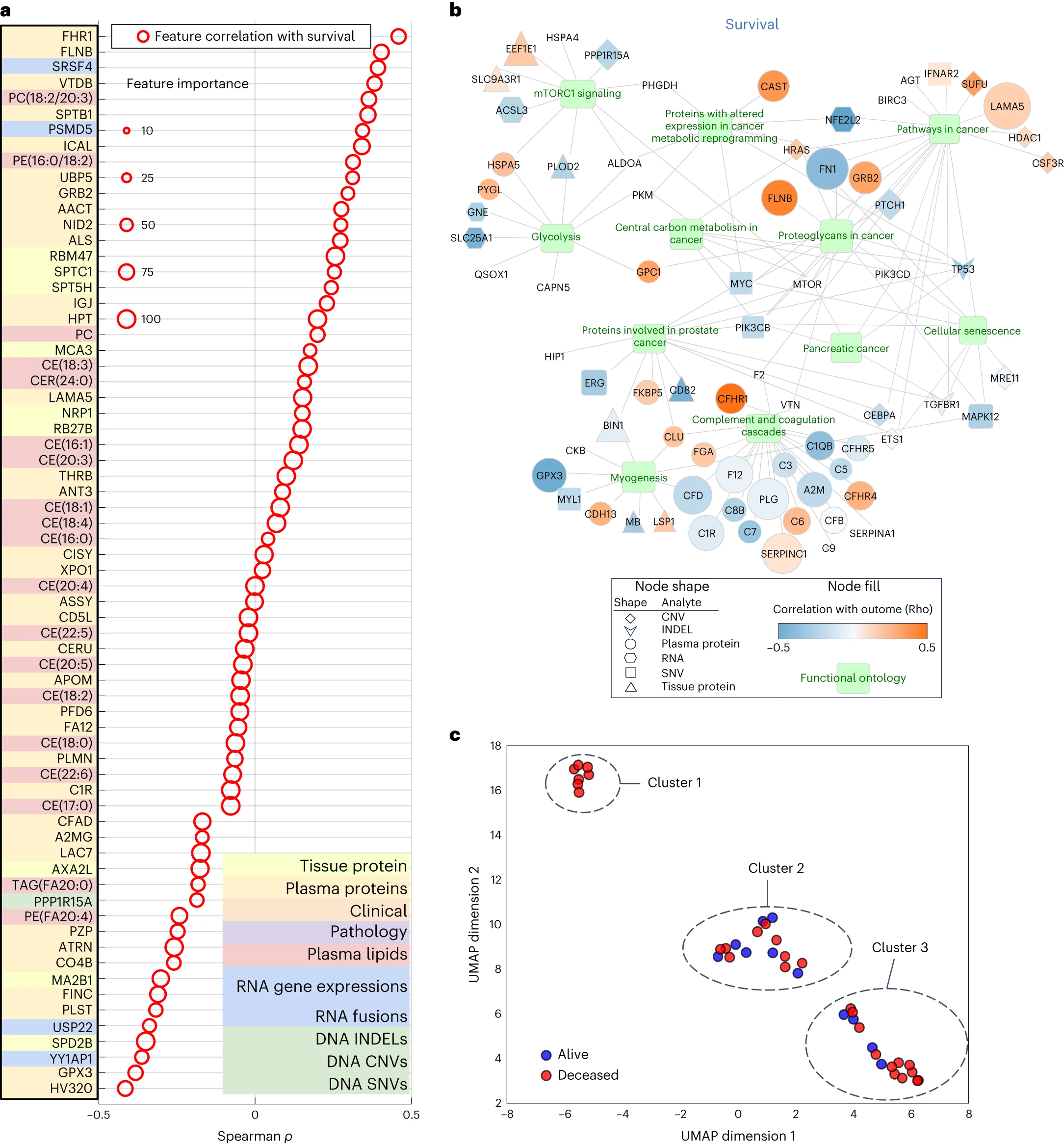
图5 多组学模型和聚类中主要特征的生物学相关性
结论8:
预测疾病生存的简约多组学模型
前面提到,多组学模型呈现了各个分析层面之间的互补性。在这一部分中,研究人员利用递归特征消除策略,从完整的多组学特征集中逐步“消除”最不相关的特征,最终形成一个简约模型——该模型仅需589个多组学特征,但准确性和阳性预测值均达到0.85,具有与更大、更复杂的模型相似的预测性能
该简约模型主要受血浆脂质和RNA的影响,而在模型形成后的迭代过程中,这些影响基本保持稳定。这表明仅通过血浆(蛋白质或脂质)的检测,就有可能做出关于胰腺手术的决策。
通过评估特定有限的分析物组合和特征集,研究者发现可以应用于简约模型的分析物,这些分析物具有标准可用性(病理标本或临床数据,包括外科病理学)或易于获取(血浆脂质或蛋白质)的特点。这些简约模型包括从临床、外科病理学和计算病理学分析物中学到的特征、所有血浆分析物(脂质组学和蛋白质)学到的特征,以及从临床、计算病理学和血浆分析物中学到的特征。这些简约模型的准确性和阳性预测值与从完整特征集中学到的模型相似,为最终实现精准癌症医学的全球化提供了潜在的可能性。
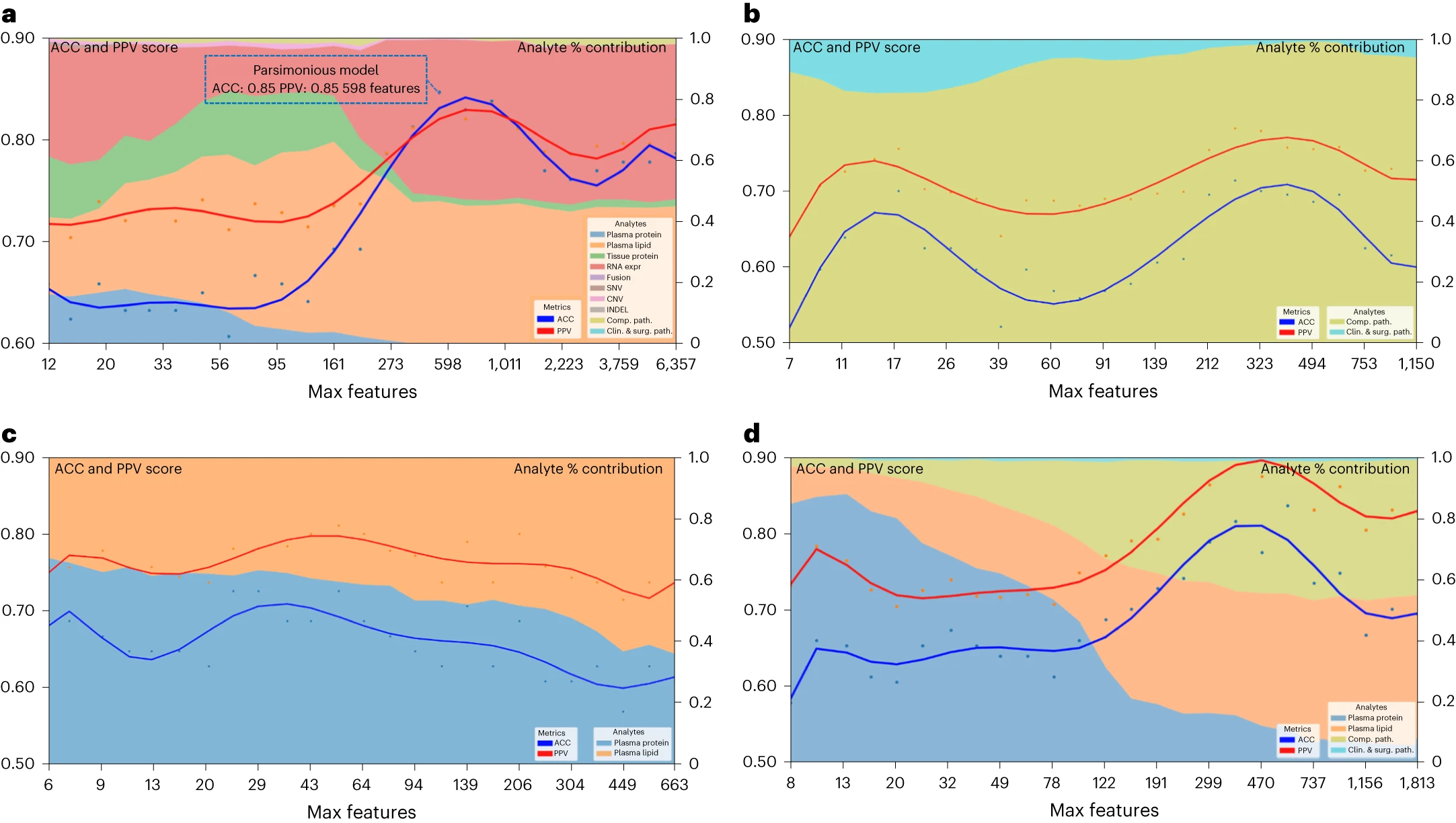
图6 简约多组学模型的性能和分析物对疾病生存的贡献 a:所有多组学特征和完整数据集的简约模型。蓝色虚线框表示拐点处的简约模型;b:仅包含临床病理学、手术病理学和计算病理学分析的模型;c:仅包含所有血浆分析(脂质组学和蛋白质)的模型;d:仅包含所有临床病理学、手术病理学、计算病理学和血浆分析(脂质组学和蛋白质)的模型。左 y 轴显示准确性和阳性预测值(PPV)分数:多组学模型性能随着限制模型训练中最大可选择特征的数量的减少而变化。x 轴显示每个减少步骤的最大特征数。右 y 轴显示分析物贡献百分比(%):每种分析物在每个减少步骤中的聚合绝对特征权重贡献。
结论9:
验证预测存活率的RNA标记物
研究人员对试点队列中的57个样本进行了全转录组测序和分析,利用L1标准化的随机森林模型,他们筛选出了显著预测胰腺癌患者生存的RNA基因转录本,并分别构建了与良好生存相关的基因标志(40个基因)和与不良生存相关的基因标志(39个基因)。这两个标志在包含177名胰腺癌患者的独立数据集中进行验证,用于分层分化DS。
与在本研究中与不良预后相关的基因表达标志高得分者,其在验证集中也显示出不良DS;而与在本研究中与良好预后相关的基因表达标志高得分者,其在验证集中趋向于改善DS。
此外,对这两个标志中使用的RNA转录本进行基因集富集分析后,研究人员发现了与PDAC抗药性和治疗靶向相关的多条显著通路。这些数据进一步验证了RNA表达的临床相关性。
结论10:
验证预测存活率的多组学模型
为了进一步验证前述单组学、多组学和简化组学分析用于DS预测的性能,研究人员评估了它们在TCGA数据集上的预测性能。结果显示,多组学简明模型在TCGA数据集上同样表现出较好的准确性和PPV,验证了其泛化能力。同时,在另一个独立的含有组织蛋白数据的验证集(JHU)上进行了验证,表明多组学方法的有效性。
此外,虽然一个仅以组织蛋白质为单组学分析的模型在JHU队列1中的准确度和PPV为0.56和0.53,但DNA、RNA和临床分析的添加提高了模型的预测性能,并验证了多组学的方法。
总 结
总的来说,该研究利用先进的机器学习模型分析了包括6363个临床和多组学分子特征的数据集,以准确预测胰腺导管腺癌(PDAC)患者的疾病生存情况。实验证明,完整的多组学模型能够以最高的准确度预测PDAC疾病生存情况,而血浆蛋白是疾病生存情况的顶级单组学预测因。
重要的是,该平台内的所有单组学和多组学建模作为临床预测工具的表现均明显优于癌抗原19-9(CA19-9),这对于癌症研究和个体化治疗具有潜在的重要意义。
尽管该研究有一些限制(如样本量较小和不包括所有可能的分析类型),但研究人员强调了这一方法的可扩展性和灵活性。未来,该领域的工作将包括对更大样本量的研究、纳入更多类型的分析、以及对肿瘤异质性和微环境的更深入分析。
文末彩蛋
该平台为何取名为 Molecular Twin?
在该研究的 Research Briefing 中,作者提到,“我们将这个平台命名为 Molecular Twin(‘分子双胞胎’),是为这样一个美好愿景:最终将存在数千个已知结果的 twins,并将帮助建立一个参考数据库,用于预测性地将新的 twins 与最佳治疗相匹。”
他们表示,尽管这一针对PDAC患者的研究使用了 Molecular Twin,但这一方法和平台有可能以与肿瘤无关的方式应用于所有癌症类型。
使用多组学方法助力疾病诊疗潜力无限,我们也期待这一天的到来!
文章链接:
https://www.nature.com/articles/s43018-023-00697-7


